Friends Who Are Unrelated Share A Surprising Amount Of DNA
Friends really are the family you choose
By Alexandra Ossola | Published Jul 16, 2014 6:21 PM EDT

You and your best friend have a lot in common: your favorite food, your taste in music, maybe your hometown. But a new study finds that your similarities may even extend to a genetic level.
The researchers, James Fowler of University of California San Diego and Nicholas Christakis of Yale University, used data collected during the famous Framingham Heart Study , running since 1948 in the small town in Massachusetts. When participants shared their DNA with researchers for the study, they shared lots of other information, too, including who they hang out with. “Because the study started in a small community, many people that were named as friends, also happened to be involved in the study,” Fowler explained to the BBC . He and Christakis looked at almost 2,000 participants and identified about 1,400 pairs of friends.
The findings , published this week in the Proceedings of the National Academy of Sciences , show that people share 0.1 percent more DNA with their friends than with perfect strangers. That’s about the same genetic similarity you share with your fourth cousin.
So why might this be the case? The study authors had a few theories. Maybe people with similar genes seek out similar environments and then meet others like them. Or, people who share DNA could have comparable skill sets, so they work together better over long periods of evolutionary time.
The study has a few limitations. For one, Fowler’s team didn’t look across the entire human genome–they compared only about 500,000 of each person’s three billion DNA base-pairs. Even though the researchers excluded anyone who was related in any way, Framingham’s population is made up mostly of descendants of Italian and Irish immigrants, so the genetic variation may not be large enough to make a broader conclusion. Evan Charney , a professor of public policy at Duke University, said that, to maintain the study’s integrity, the researchers could only study a population in which individuals are completely unrelated to one another, which is admittedly very difficult to find. Rory Bowden, a statistician at the Wellcome Trust Centre for Human Genetics in Oxford, also had reservations about how countries of origin could affect the communities that people seek out, such as church groups and cultural associations, which would align people with similar genetics.
But others, including the researchers, stand by the conclusions. Findings such as this, Fowler notes, could influence theories about how altruism has developed over evolutionary time.
Of course, Fowler and Christakis don’t have all the information yet. Interestingly, they found that the biggest genetic similarities were found in friends’ sense of smell. They’re not quite sure why that would be the case, but future studies may help them sniff out the answers.
Like science, tech, and DIY projects?
Sign up to receive Popular Science's emails and get the highlights.

Provide details on what you need help with along with a budget and time limit. Questions are posted anonymously and can be made 100% private.

Studypool matches you to the best tutor to help you with your question. Our tutors are highly qualified and vetted.

Your matched tutor provides personalized help according to your question details. Payment is made only after you have completed your 1-on-1 session and are satisfied with your session.
- Homework Q&A
- Become a Tutor
All Subjects
Mathematics
Programming
Health & Medical
Engineering
Computer Science
Foreign Languages

Access over 20 million homework & study documents
Kami export josue santiago unfriended case study student guide 1.

Sign up to view the full document!

24/7 Homework Help
Stuck on a homework question? Our verified tutors can answer all questions, from basic math to advanced rocket science !

Similar Documents
working on a homework question?
Studypool is powered by Microtutoring TM
Copyright © 2024. Studypool Inc.
Studypool is not sponsored or endorsed by any college or university.
Ongoing Conversations

Access over 20 million homework documents through the notebank
Get on-demand Q&A homework help from verified tutors
Read 1000s of rich book guides covering popular titles

Sign up with Google
Sign up with Facebook
Already have an account? Login
Login with Google
Login with Facebook
Don't have an account? Sign Up
Short videos can be used as case studies by stopping at appropriate time points and asking questions to cue student attention, encourage critical thinking, and make the students part of the story.
This video case study focuses on a real-world problem: identifying genetic mutation(s) associated with diseases. It is appropriate for a range of biology courses, including AP/IB Biology and college-level introductory biology.
Student Learning Targets
- Formulate a hypothesis to explain how a mutation in a gene would affect the function of a cell and an organism.
- Describe the possible steps involved in identifying a disease-causing gene mutation in a patient.
- Predict how replacing a mutated gene with a functioning copy of that gene will affect the phenotype of a cell and/or organism.
- Explain how the identification of disease-causing mutations can be used to develop medical treatments.
Prior Knowledge
Students should be familiar with the definition of mutations and how genetic mutations can be inherited. Students should also be familiar with the process of gene expression.
Background Information
- Most human traits, including diseases, are influenced by multiple genes. It is difficult to develop genetic therapies for diseases caused by variations or mutations in many genes (multigene diseases). However, a relatively small number of genetic diseases are caused by mutations in single genes (single-gene diseases). It’s more feasible to identify mutations for single-gene diseases and develop effective therapies.
- Retinitis pigmentosa (RP) is a progressive visual disease that causes photoreceptor cells in the retina to die. It is caused by mutations in any one of several different genes. Mutations in any one of these genes are sufficient to cause RP.
- Many mutations that cause RP have been identified. When scientists tested the DNA of the patient featured in this video, Sam, they did not find any of these known mutations.
- Scientists determined that Sam’s DNA contains a previously unknown mutation that affects tRNA structure and function. They then discovered that two other patients with RP have mutations in the same gene.
- Scientists were able to confirm that the mutation affects photoreceptor cells through experiments in zebrafish, a model research organism.
The video case study is presented in eight modules, each followed by embedded questions. Educators can use the extension questions listed below for additional class discussion or written assessments.
Modules 1 and 2 establish the genetic basis of RP and describe other features of the disease. Prior to starting the video, prompt students by asking what they know about gene therapy. Following a brief discussion, start viewing the module. The first module ends by asking students questions about the inheritance pattern of RP. In the second module, students consider how genetic analysis of DNA from blood samples of related individuals could be used to identify the causative mutation.
Extension questions:
- How do our brains convert information from light entering our eyes into an image?
- What is disease? How do organisms get diseases?
- What is a genetic mutation and what are its effects?
- How is searching for a treatment for a genetic disease different from that for a disease caused by something else, such as bacteria or viruses?
Module 3 explains how comparing the DNA of relatives can identify a mutation that causes disease and how this approach has been used to identify many different mutations that cause RP. The module ends by asking students to explain how one disease can be caused by any one of many mutations.
- Why would a researcher use a pedigree to study a disease?
- Many characteristics of an organism are controlled by more than one gene. Likewise, many of the biochemical processes and pathways involve proteins produced by different genes. Explain how this relates to the existence of many different mutations that cause RP.
- Can you think of any other diseases or conditions that are caused by mutations in different genes?
Modules 4 and 5 explore how a genetic scan of Sam’s genome did not find any of the known common or rare mutations that cause RP. Instead, Sam’s DNA has a mutation that had never been identified as a possible cause of RP. These two modules end with questions asking students to interpret the results of the genetic analysis and predict the impact of the mutation affecting Sam’s tRNA function.
Modules 6 reveals how a mutation that affects tRNA function is not necessarily lethal. The mutation in Sam’s DNA reduces tRNA function but does not completely stop protein synthesis. Knowing this, students are asked what additional data they would need to collect as evidence that the mutation in Sam’s DNA causes RP.
Extension question:
- Some mutations are lethal and others are not. Explain the difference between these two types of mutations in terms of cellular function.
Modules 7 and 8 conclude the case study by demonstrating the experimental procedure used to confirm that the mutation in Sam’s DNA causes RP. By genetically engineering zebrafish, a model organism, to have a mutation in the same gene, scientists demonstrated that the mutation impacts how zebrafish respond to light. With this information, students conclude the case study by predicting how gene therapy could be used to treat RP and the outcomes for patients’ vision. The final module concludes by asking students to explain the benefits of identifying mutations that cause disease.
- In your own words, explain why researchers use model organisms such as zebrafish. Can you name any other model organisms?
- Genetic medicine is an emerging technology that holds great promise for many diseases. Are there any ethical concerns with genetic medicine as described in this case study? Explain your perspective.
- Are there any other diseases you know of that currently are or could potentially be treated or cured through genetic medicine?
- Search newspapers, magazines, or the scientific literature for diseases being treated through genetic medicine. Have there been successes? Failures?
A DNA Case Study: Revealing a Misattributed Parentage Event with DNA
As DNA testing for genealogy becomes increasingly popular, more individuals are using the tool to examine and confirm their family trees. However, as more people are tested and comparing DNA to their paper trail, more people are discovering that their genetic ancestry is not what they expected it to be.
The Genetic Genealogy Standards were created to help educate people about the possible outcomes and limitations of genetic genealogy testing. One of the possible outcomes is misattributed parentage, or the discovery that a supposed genealogical ancestor is not in one’s genetic line. Many correctly point out that a misattributed ancestor is still be a social ancestor firmly rooted in one’s social tree, although it is potentially important to know when an ancestor is not one’s genetic ancestor.
Here we will look at a real-life example of how DNA testing uncovered a family secret nearly 100 years old.
The Clancy Study
The Clancys are a family of interest for me, and I wanted to test several descendants of the immigrant family using autosomal DNA. This would confirm the family tree, find other people related through this family, and potentially give me leads back in the originating country.
Since I am not a descendant through an unbroken male line, I also wanted to find a Clancy male who would be willing to take a Y-DNA test. I could learn more about the ancient origins of the Y-DNA line, and potentially find matches that I could work together with to extend the family tree.
I had already tested two great-grandchildren of the immigrant couple – Melissa Smith and Robert Smith in FIG. 1 – and these would be a great starting point for the Clancy Study. Now I just needed to find other willing descendants of Daley and Mary (Shane) Clancy.
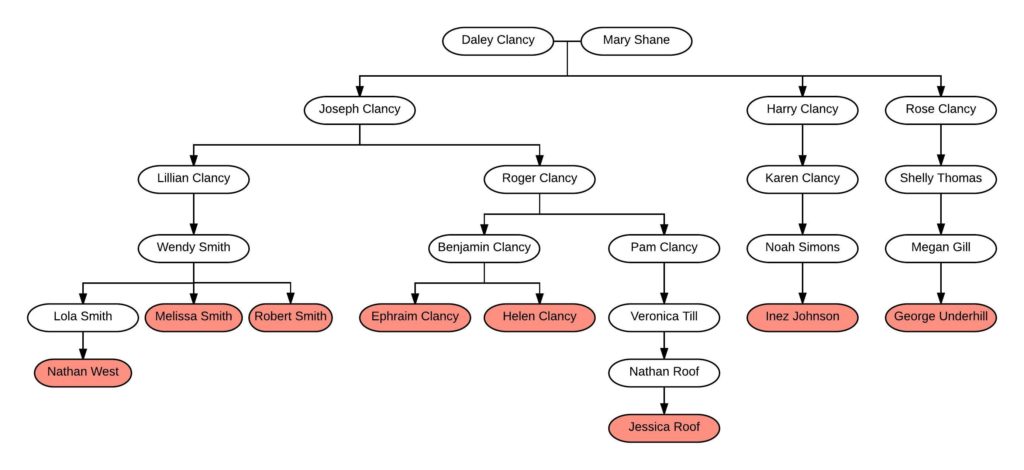
We will reference this chart often. You can click on it to enlarge it, and you might want to print it out or open it in a new tab to have it handy.
Finding a Test Subject
Before DNA hit the scene, I’d spent a great deal of time researching the Clancy family and documenting the descendants. In the late 1990’s and early 2000’s I wrote to many descendants and received magnificent letters back describing what they knew about the family, each containing a tidbit of lore or history about the Clancys. Many of that older generation are now gone, and these letters are invaluable treasures.
Because I’d researched the family, it was an easy matter to identify a living male Clancy descendant – Ephraim Clancy. Once I contacted him, he was happy to take a Y-DNA and atDNA test. His sister – Helen Clancy – was also happy to take an atDNA test.
Referring to FIG. 1 again, you can see that Melissa Smith and Robert Smith are second cousins to Ephraim Clancy and Helen Clancy. This was perfect, as there was a guarantee that these second cousins would share DNA (to date, no one has ever proven a second cousin or closer relationship that does NOT share a detectable amount of DNA).
Other Clancy Descendants Already in the Database
Additionally, testing Melissa and Robert Smith had revealed other descendants of Daley and Mary (Shane) Clancy already in the testing company database. For example, Inez Johnson and George Underhill are descendants through two other children of Daley and Mary as shown in FIG. 1.
The family tree shows that Melissa Smith and Robert Smith are third cousins to Inez Johnson and George Underhill. Both Melissa and Robert share DNA with Inez and George at an expected level. According to the 2016 data from the Shared cM Project , 3C has an average of 79 cM shared with a range from 0 to 200 cM or so. All the 3C relationships fall nicely into that range:
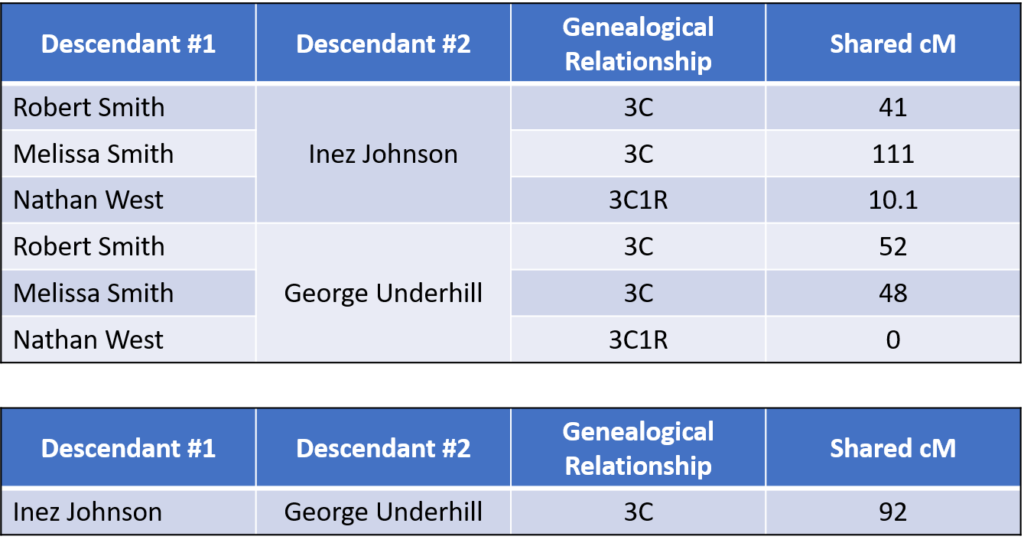
With these results, I have DNA evidence that potentially supports documentation that Joseph, Harry, and Rose Clancy are siblings and children of Daley and Mary (Shane) Clancy.
Now let’s bring in the DNA test results from Ephraim and Helen.
The Results
Siblings Ephraim and Helen share about 2,700 cM with plenty of fully identical regions, making them full siblings as expected.
However, when comparing their shared DNA to their extended relatives, we discovered the following:
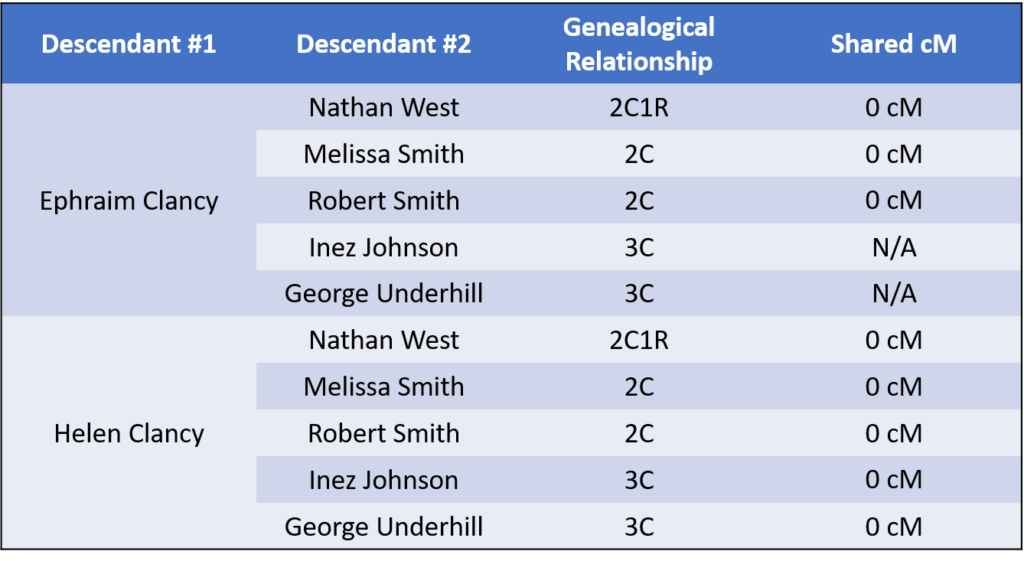
This should raise many red flags, considering that there are a total of four different 2C relationships here that do not share DNA. As of today (March 13, 2017), no second cousin relationship (or closer) has ever been proven to share no DNA. Accordingly, there appears to be a misattributed parentage event in this tree somewhere.
Narrowing in on the Misattributed Parentage Event
A final piece of information helped pinpoint which generation the MPE likely occurred. While Ephraim and Helen were testing, I discovered that their close relative, Jessica Roof, had recently tested.
As expected, Jessica is closely related to Nathan, Melissa, and Robert. For example, the 2C2R relationships are almost exactly the same as the average (81 cM), and the 3C1R relationship is within the normal range (0-156 cM, average 56 cM):
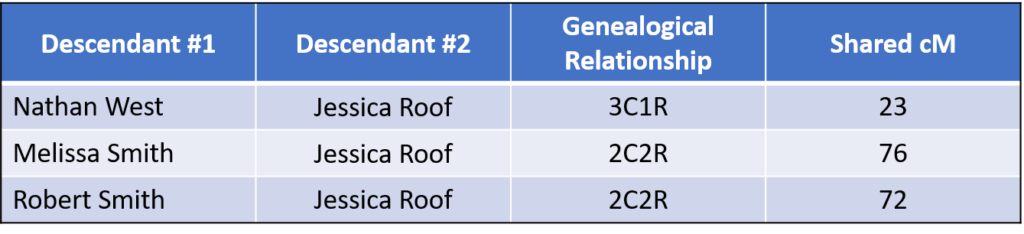
As shown in FIG. 1,Jessica is a 1C2R (first cousin twice removed) to Ephraim and Helen, a very close relationship. The results of the DNA testing shows that Jessica shares the following amounts of DNA with her cousins Ephraim and Helen:
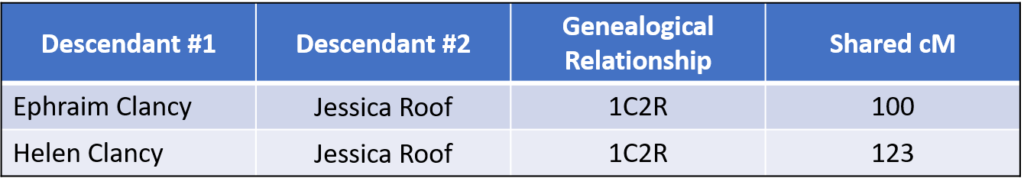
This was somewhat surprising. According to the 2016 data from the Shared cM Project , the average for 1C2R is 235 cM with a range of 27 to 413 cM. Although both numbers were well within the range, they were both almost half of the average amount.
OK. Now, with this information and before you peek down below, can you come up with a scenario that explains the DNA results we’ve seen? Can you determine where the misattributed parentage event likely occurred? Scroll down when you are ready for my hypothesis, and see whether you agree.
Identifying a Misattributed Parentage Event
If we hypothesize that Benjamin and Pam were maternal half-siblings (with Pam being the daughter of Roger Clancy and his wife but Benjamin being a child of the wife and another man) then ALL the results could fit within that model. If Benjamin and Pam are maternal half-siblings, for example, that would mean that Ephraim and Helen are not biologically Clancys. Instead of grandfather Roger Clancy, another name should be there in the genetic family tree for them. Pam, however, is likely the child of Roger Clancy as described below.
The maternal half-sibling theory resolves several other concerns:
- For example, this theory would explain why Ephraim and Helen don’t share DNA with any of their 2Cs or 3Cs (i.e., Melissa, Robert, Inez, and George). Ephraim and Helen are related to all of those individuals only through Roger Clancy, according to the tree. Because Jessica Roof is also related to all of those individuals only through Roger Clancy, her line appears to be correct (because she DOES share DNA with Melissa, Robert, and Nathan, for example).
- The theory also explains why Jessica Roof shares DNA with Ephraim and Helen, potentially at 50% of the average. If Benjamin and Pam were maternal half siblings, then Ephraim and Helen would be half 1C2R to Jessica. This is not as clear because the shared amount fits within the broad range for 1C2R, but it fits perfectly into the model.
Thus, according to this theory, Roger Clancy had just one child – Pam Clancy – with his wife. Some years later, there was then a second child – Benjamin Clancy – born to the wife and someone else. Of course I don’t know the circumstances of this misattributed parentage event, and that is always an incredibly important consideration.
This is, of course, just the beginning of the research. I can look for additional evidence that would support my theory, both DNA evidence and documentary evidence. Additionally, I can look for the father of Benjamin Clancy using existing matches in the testing database.
(Of course, all names herein have been changed, as have a few other facts to avoid identification).
Share this:
13 responses.
Great illustration and example of how relations “should” work within fairly close relationships. Curious if the yDNA for “Ephraim Clancy” yielded any clues
Great explanation! I think a lot of times we’re running into the reverse situation – we’ve got DNA matches but we can’t find the MRCA in the related trees. I’d like to see a similar use of Ancestry DNA results that point to a tree but can’t find the MRCA – I’m suspecting, given sufficiently large trees, that we are encountering NPEs.
Nice. It’s obvious Benjamin is not who he is supposed to be. Pity there wasn’t another male Clancy descendant and you could have sorted it out with only two Y-tests.
But did you consider going back another generation or two and looking for more male Clancys? It’s the easiest and absolutely guaranteed. (You would need to triangulate with three Y-tests).
It looks like your hypothesis can be affirmed from the results that you have. Does Jessica Roof also match Inez Johnson and George Underhill (although more distant than the relationship the Smiths and Nathan West)?
Could another possibility be that Benjamin Clancy really is the biological son of Roger Clancy and that Benjamin’s children could be descended from Benjamin’s wife and another man?
This answer should be a possibility, but is not suggested by the descendant chart they used that does not show spouses (except Mary Shane). This descendant chart could be correct through Benjamin Clancy, and should not include Ephraim and Helen.
No, because then they wouldn’t be expected to share DNA with Jessica. Jessica’s results are what really pinpoint the relationships.
Yes, great example. I wanted to scroll down at once but had to restrain myself. I was glad I didn’t since I figured it out. Couldn’t these kind of puzzles not be formalized into some automated program? Then the program could come up with alternative scenarios….
Yes, this is a perfect scenario for a computer program to do some analysis and present potential anomalies for further review. Unfortunately, coming up with an exhaustive list of alternative scenarios would be quite difficult due to the number of factors involved. Nonetheless, this is exactly the type of scenario we have found quite lacking in the current genealogy software programs and the reason we are in the process of creating a new program that can handle DNA, genetics and more. We’d love to hear more about how people need to use DNA data in their research: http://www.heirloomsoftware.com
We absolutely love your blog and find a lot of your post’s to be what precisely I’m looking for. can you offer guest writers to write content for you personally? I wouldn’t mind publishing a post or elaborating on most of the subjects you write about here. Again, awesome site!|
Regarding MPE’s. I purchased your guide to DNA testing, and am gradually understanding the contents. For two years, I have been seeking the biological father of a friend with only DNA reports (uploaded to GEDmatch); distant cousins as matches; and a couple of weak clues to work with. I have charted numerous mirror trees, and now have a theory that requires the cooperation of a possible half-sibling. This places me in the role of the “skunk at the family picnic” as I attempt to get suspected close relatives to take a DNA test which might reveal marital infidelity. While a genealogy question begins as an intriguing, enjoyable puzzle, the fun is likely to transform into a harsher reality when you come closer to the correct answer. You may find yourself in that position with the “Clancy” family even though the MPE happened over a century ago. I am looking for ethical guidelines and advice on how to contact people who are suspected to be related to the biological parent of a client. I am quite good at solving puzzles; but when it comes to uncovering family secrets, success may come with a bitter taste. I am a descendant of the Mohawk Valley Bellinger’s, and would like to discuss that with you in a more appropriate forum. Thank you for your good work.
An interesting study I have to say. It would be nice to explain some details but I hope that essay writing app at https://essayyoda.com/essay-writing-app/ would help me with this. It’s always useful to get professional help in the pocket.
https://essayyoda.com/essay-writing-app/ is a good education site
Comments are closed.
Privacy Overview

- school Campus Bookshelves
- menu_book Bookshelves
- perm_media Learning Objects
- login Login
- how_to_reg Request Instructor Account
- hub Instructor Commons
- Download Page (PDF)
- Download Full Book (PDF)
- Periodic Table
- Physics Constants
- Scientific Calculator
- Reference & Cite
- Tools expand_more
- Readability
selected template will load here
This action is not available.
8.1: Case Study: Genes and Inheritance
- Last updated
- Save as PDF
- Page ID 16760

- Suzanne Wakim & Mandeep Grewal
- Butte College
Case Study: Cancer in the Family
People tend to look similar to their biological parents, as illustrated by the family tree in Figure \(\PageIndex{1}\). But, you can also inherit traits from your parents that you can’t see. Rebecca becomes very aware of this fact when she visits her new doctor for a physical exam. Her doctor asks several questions about her family's medical history, including whether Rebecca has or had relatives with cancer. Rebecca tells her that her grandmother, aunt, and uncle, who have all passed away, all had cancer. They all had breast cancer, including her uncle, and her aunt additionally had ovarian cancer. Her doctor asks how old they were when they were diagnosed with cancer. Rebecca is not sure exactly, but she knows that her grandmother was fairly young at the time, probably in her forties.

Rebecca’s doctor explains that while the vast majority of cancers are not due to inherited factors, a cluster of cancers within a family may indicate that there are mutations in certain genes that increase the risk of getting certain types of cancer, particularly breast and ovarian cancer. Some signs that cancers may be due to these genetic factors are present in Rebecca’s family, such as cancer with an early age of onset (e.g. breast cancer before age 50), breast cancer in men, and breast cancer and ovarian cancer within the same person or family.
Based on her family medical history, Rebecca’s doctor recommends that she see a genetic counselor because these professionals can help determine whether the high incidence of cancers in her family could be due to inherited mutations in their genes. If so, they can test Rebecca to find out whether she has the particular variations of these genes that would increase her risk of getting cancer.
When Rebecca sees the genetic counselor, he asks how her grandmother, aunt, and uncle with cancer are related to her. She says that these relatives are all on her mother’s side — they are her mother’s mother and siblings. The genetic counselor records this information in the form of a specific type of family tree, called a pedigree, indicating which relatives had which type of cancer and how they are related to each other and to Rebecca. He also asks her ethnicity. Rebecca says that her family, on both sides, are Ashkenazi Jews, meaning Jews whose ancestors came from central and eastern Europe. “But what does that have to do with anything?” she asks. The counselor tells Rebecca that mutations in two tumor-suppressor genes called BRCA1 and BRCA2, located on chromosome 17 and 13, respectively, are particularly prevalent in people of Ashkenazi Jewish descent and greatly increase the risk of getting cancer. About 1 in 40 Ashkenazi Jewish people have one of these mutations, compared to about 1 in 800 in the general population. Her ethnicity, along with the types of cancer, age of onset, and the specific relationships between her family members who had cancer indicate to the counselor that she is a good candidate for genetic testing for the presence of these mutations.
Rebecca says that her 72-year-old mother never had cancer, and nor had many other relatives on that side of the family, so how could the cancers be genetic? The genetic counselor explains that the mutations in the BRCA1 and BRCA2 genes, although dominant, are not inherited by everyone in a family. Also, even people with mutations in these genes do not necessarily get cancer — the mutations simply increase their risk of getting cancer. For instance, 55 to 65% of women with a harmful mutation in the BRCA1 gene will get breast cancer before age 70, compared to 12% of women in the general population who will get breast cancer sometime over the course of their lives.
Rebecca is not sure she wants to know whether she has a higher risk of cancer. The genetic counselor understands her apprehension but explains that if she knows that she has harmful mutations in either of these genes, her doctor will screen her for cancer more often and at earlier ages. Therefore, any cancers she may develop are likely to be caught earlier when they are often much more treatable. Rebecca decides to go through with the testing, which involves taking a blood sample, and nervously waits for her results.
Chapter Overview: Genetics
At the end of this chapter, you will find out Rebecca ’s test results. By then, you will have learned how mutations in genes such as BRCA1 and BRCA2 can be passed down and cause disease. Especially, you will learn about:
- How Gregor Mendel discovered the laws of inheritance for certain types of traits.
- The science of heredity, known as genetics, and the relationship between genes and traits.
- Simple and more complex inheritance of some human traits.
- Genetic Disorders.
As you read this chapter, keep Rebecca’s situation in mind and think about the following questions:
- What do the BRCA1 and BRCA2 genes normally do? How can they cause cancer?
- Are BRCA1 and BRCA2 considered linked genes? And are they on autosomes or sex chromosomes?
- After learning more about pedigrees, draw the pedigree for cancer in Rebecca’s family. Use the pedigree to help you think about why it is possible that her mother does not have one of the BRCA gene mutations, even if her grandmother, aunt, and uncle did have it.
- Why do you think certain gene mutations are prevalent in certain ethnic groups?
Attributions
- Caelius and Valerius family tree by Ann Martin , licensed CC BY 2.0 via Flickr
- Text adapted from Human Biology by CK-12 licensed CC BY-NC 3.0
Laboratory Analysis of Biological Evidence and the Role of DNA in Sexual Assault Investigations
This comprehensive module explores the complex role of DNA in a sexual assault investigation, including the alternative sources of DNA evidence and their potential significance or impact on a sexual assault investigation. A number of resources and tools are provided, along with a series of complex and interactive case examples.
Estimated time for completion: 32 hours
Download Course Outline | Download Document
EVAWI uses cookies on our website to improve your experience.
By continuing to use this site, you consent to our use of cookies. For more information, please see our Privacy Policy.
Note: on the next page you will be asked for an email address. Make sure to fill the field out using the email address of the person who should be enrolled in the course. Use your own email address if you are purchasing this course for yourself.
If you need receipts for courses you are purchasing for other people, please contact [email protected] .
Continue to checkout
Want to create or adapt books like this? Learn more about how Pressbooks supports open publishing practices.
5.1 Case Study: Genes and Inheritance
Created by: CK-12/Adapted by Christine Miller
Case Study: Cancer in the Family
People tend to carry similar traits to their biological parents, as illustrated by the family tree. Beyond just appearance, you can also inherit traits from your parents that you can’t see.
Rebecca becomes very aware of this fact when she visits her new doctor for a physical exam. Her doctor asks several questions about her family medical history, including whether Rebecca has or had relatives with cancer. Rebecca tells her that her grandmother, aunt, and uncle — who have all passed away — had cancer. They all had breast cancer, including her uncle, and her aunt also had ovarian cancer. Her doctor asks how old they were when they were diagnosed with cancer. Rebecca is not sure exactly, but she knows that her grandmother was fairly young at the time, probably in her forties.
Rebecca’s doctor explains that while the vast majority of cancers are not due to inherited factors, a cluster of cancers within a family may indicate that there are mutations in certain genes that increase the risk of getting certain types of cancer, particularly breast and ovarian cancer. Some signs that cancers may be due to these genetic factors are present in Rebecca’s family, such as cancer with an early age of onset (e.g., breast cancer before age 50), breast cancer in men, and breast cancer and ovarian cancer within the same person or family.
Based on her family medical history, Rebecca’s doctor recommends that she see a genetic counselor, because these professionals can help determine whether the high incidence of cancers in her family could be due to inherited mutations in their genes. If so, they can test Rebecca to find out whether she has the particular variations of these genes that would increase her risk of getting cancer.
When Rebecca sees the genetic counselor, he asks how her grandmother, aunt, and uncle with cancer are related to her. She says that these relatives are all on her mother’s side — they are her mother’s mother and siblings. The genetic counselor records this information in the form of a specific type of family tree, called a pedigree, indicating which relatives had which type of cancer, and how they are related to each other and to Rebecca.
He also asks her ethnicity. Rebecca says that her family on both sides are Ashkenazi Jews (Jews whose ancestors came from central and eastern Europe). “But what does that have to do with anything?” she asks. The counselor tells Rebecca that mutations in two tumor-suppressor genes called BRCA1 and BRCA2 , located on chromosome 17 and 13, respectively, are particularly prevalent in people of Ashkenazi Jewish descent and greatly increase the risk of getting cancer. About one in 40 Ashkenazi Jewish people have one of these mutations, compared to about one in 800 in the general population. Her ethnicity, along with the types of cancer, age of onset, and the specific relationships between her family members who had cancer, indicate to the counselor that she is a good candidate for genetic testing for the presence of these mutations.
Rebecca says that her 72-year-old mother never had cancer, nor had many other relatives on that side of the family. How could the cancers be genetic? The genetic counselor explains that the mutations in the BRCA1 and BRCA2 genes, while dominant, are not inherited by everyone in a family. Also, even people with mutations in these genes do not necessarily get cancer — the mutations simply increase their risk of getting cancer. For instance, 55 to 65 per cent of women with a harmful mutation in the BRCA1 gene will get breast cancer before age 70, compared to 12 per cent of women in the general population who will get breast cancer sometime over the course of their lives.
Rebecca is not sure she wants to know whether she has a higher risk of cancer. The genetic counselor understands her apprehension, but explains that if she knows that she has harmful mutations in either of these genes, her doctor will screen her for cancer more often and at earlier ages. Therefore, any cancers she may develop are likely to be caught earlier when they are often much more treatable. Rebecca decides to go through with the testing, which involves taking a blood sample, and nervously waits for her results.
Chapter Overview: Genetics
At the end of this chapter, you will find out Rebecca’s test results. By then, you will have learned how traits are inherited from parents to offspring through genes, and how mutations in genes such as BRCA1 and BRCA2 can be passed down and cause disease. Specifically, you will learn about:
- The structure of DNA.
- How DNA replication occurs.
- How DNA was found to be the inherited genetic material.
- How genes and their different alleles are located on chromosomes.
- The 23 pairs of human chromosomes, which include autosomal and sex chromosomes.
- How genes code for proteins using codons made of the sequence of nitrogen bases within RNA and DNA.
- The central dogma of molecular biology, which describes how DNA is transcribed into RNA, and then translated into proteins.
- The structure, functions, and possible evolutionary history of RNA.
- How proteins are synthesized through the transcription of RNA from DNA and the translation of protein from RNA, including how RNA and proteins can be modified, and the roles of the different types of RNA.
- What mutations are, what causes them, different specific types of mutations, and the importance of mutations in evolution and to human health.
- How the expression of genes into proteins is regulated and why problems in this process can cause diseases, such as cancer.
- How Gregor Mendel discovered the laws of inheritance for certain types of traits.
- The science of heredity, known as genetics, and the relationship between genes and traits.
- How gametes, such as eggs and sperm, are produced through meiosis.
- How sexual reproduction works on the cellular level and how it increases genetic variation.
- Simple Mendelian and more complex non-Mendelian inheritance of some human traits.
- Human genetic disorders, such as Down syndrome, hemophilia A, and disorders involving sex chromosomes.
- How biotechnology — which is the use of technology to alter the genetic makeup of organisms — is used in medicine and agriculture, how it works, and some of the ethical issues it may raise.
- The human genome, how it was sequenced, and how it is contributing to discoveries in science and medicine.
As you read this chapter, keep Rebecca’s situation in mind and think about the following questions:
- BCRA1 and BCRA2 are also called Breast cancer type 1 and 2 susceptibility proteins. What do the BRCA1 and BRCA2 genes normally do? How can they cause cancer?
- Are BRCA1 and BRCA2 linked genes? Are they on autosomal or sex chromosomes?
- After learning more about pedigrees, draw the pedigree for cancer in Rebecca’s family. Use the pedigree to help you think about why it is possible that her mother does not have one of the BRCA gene mutations, even if her grandmother, aunt, and uncle did have it.
- Why do you think certain gene mutations are prevalent in certain ethnic groups?
Attributions
Figure 5.1.1
Family Tree [all individual face images] from Clker.com used and adapted by Christine Miller under a CC0 1.0 public domain dedication license (https://creativecommons.org/publicdomain/zero/1.0/).
Figure 5.1.2
Rebecca by Kyle Broad on Unsplash is used under the Unsplash License (https://unsplash.com/license).
Wikipedia contributors. (2020, June 27). Ashkenazi Jews. In Wikipedia. https://en.wikipedia.org/w/index.php?title=Ashkenazi_Jews&oldid=964691647
Wikipedia contributors. (2020, June 22). BRCA1. In Wikipedia . https://en.wikipedia.org/w/index.php?title=BRCA1&oldid=963868423
Wikipedia contributors. (2020, May 25). BRCA2. In Wikipedia. https://en.wikipedia.org/w/index.php?title=BRCA2&oldid=958722957
Human Biology Copyright © 2020 by Christine Miller is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License , except where otherwise noted.
Share This Book
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- My Account Login
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Open access
- Published: 20 February 2024
Cases of trisomy 21 and trisomy 18 among historic and prehistoric individuals discovered from ancient DNA
- Adam Benjamin Rohrlach 1 , 2 ,
- Maïté Rivollat 1 , 3 , 4 , 5 ,
- Patxuka de-Miguel-Ibáñez ORCID: orcid.org/0000-0003-3957-3378 6 , 7 , 8 ,
- Ulla Moilanen ORCID: orcid.org/0000-0002-4213-4061 9 ,
- Anne-Mari Liira 10 ,
- João C. Teixeira ORCID: orcid.org/0000-0001-6417-4702 11 , 12 , 13 , 14 ,
- Xavier Roca-Rada ORCID: orcid.org/0000-0001-7502-6270 12 ,
- Javier Armendáriz-Martija 15 ,
- Kamen Boyadzhiev 16 ,
- Yavor Boyadzhiev 16 ,
- Bastien Llamas ORCID: orcid.org/0000-0002-5550-9176 12 , 13 , 17 , 18 ,
- Anthi Tiliakou 1 ,
- Angela Mötsch 1 , 19 ,
- Jonathan Tuke 2 ,
- Eleni-Anna Prevedorou 20 ,
- Naya Polychronakou-Sgouritsa 21 ,
- Jane Buikstra ORCID: orcid.org/0000-0003-0206-0165 22 ,
- Päivi Onkamo 9 , 23 ,
- Philipp W. Stockhammer ORCID: orcid.org/0000-0003-4702-9372 1 , 19 , 24 ,
- Henrike O. Heyne 25 , 26 , 27 ,
- Johannes R. Lemke ORCID: orcid.org/0000-0002-4435-6610 28 , 29 ,
- Roberto Risch ORCID: orcid.org/0000-0001-8534-5806 30 ,
- Stephan Schiffels ORCID: orcid.org/0000-0002-1017-9150 1 ,
- Johannes Krause ORCID: orcid.org/0000-0001-9144-3920 1 ,
- Wolfgang Haak ORCID: orcid.org/0000-0003-2475-2007 1 &
- Kay Prüfer ORCID: orcid.org/0000-0001-6242-3058 1
Nature Communications volume 15 , Article number: 1294 ( 2024 ) Cite this article
24k Accesses
1 Citations
1106 Altmetric
Metrics details
Aneuploidies, and in particular, trisomies represent the most common genetic aberrations observed in human genetics today. To explore the presence of trisomies in historic and prehistoric populations we screen nearly 10,000 ancient human individuals for the presence of three copies of any of the target autosomes. We find clear genetic evidence for six cases of trisomy 21 (Down syndrome) and one case of trisomy 18 (Edwards syndrome), and all cases are present in infant or perinatal burials. We perform comparative osteological examinations of the skeletal remains and find overlapping skeletal markers, many of which are consistent with these syndromes. Interestingly, three cases of trisomy 21, and the case of trisomy 18 were detected in two contemporaneous sites in early Iron Age Spain (800-400 BCE), potentially suggesting a higher frequency of burials of trisomy carriers in those societies. Notably, the care with which the burials were conducted, and the items found with these individuals indicate that ancient societies likely acknowledged these individuals with trisomy 18 and 21 as members of their communities, from the perspective of burial practice.
Similar content being viewed by others

Network of large pedigrees reveals social practices of Avar communities

Genome assembly in the telomere-to-telomere era

Genome-wide association studies
Introduction.
The question of how past societies were affected by and responded to disease has been a focal point of biological anthropology for decades 1 , 2 . The physical and mental manifestations of disease can affect the ability of individuals to function in their day-to-day life, and hence can have a direct impact on the community, shaping its response to the illness 3 . The study of pathologies in human skeletal remains from archaeological contexts has been one of the most direct approaches to understanding diseases and their effect on past communities. However, osteological examinations are limited to detecting pathologies that manifest in skeletal tissue and the causes of skeletal lesions are often not simple to diagnose, as many can be expressions of different ailments. Additionally, rare diseases remain under-represented in bioarchaeological literature due to a combination of taphonomic, methodological and public visibility factors 4 , 5 . Hence, a multidisciplinary approach, such as integrating genetic, anthropological and archaeological data, can provide new insights into disease, and how it might have been perceived in past societies.
Individuals with chromosomal trisomies carry three copies of a chromosome in their cells, instead of two, following an incomplete separation (disjunction) during meiosis 6 . Except in cases of trisomy mosaicism (where only some cells are affected), or partial trisomy (where only a part of a chromosome is duplicated), individuals with full autosomal trisomies rarely survive until birth. Excluding the exceedingly rare instances of live births with trisomy 22, only three types of full autosomal trisomies can be non-fatal: trisomy 21 (Down syndrome), trisomy 18 (Edwards syndrome) and trisomy 13 (Patau syndrome). However, individuals carrying trisomy 13 or trisomy 18 rarely survive into childhood without modern medical intervention 6 , 7 .
Each case of trisomy 13, 18 and 21 is associated with congenital and sometimes severe physical and neurodevelopmental symptoms. Common symptoms for all three trisomies, such as microcephaly and brachycephaly 8 , will likely also have been recognisable to past societies. Specifically, the external physical manifestations of Down syndrome usually develop with age and can lead to a missed diagnosis of the syndrome 9 . Further, it has been shown that certain regions in the genome are responsible for eight phenotypes, leading to further phenotypic variability 10 . Few documented cases of trisomies are known from history and only a handful of cases of Down syndrome have been suggested or described in anthropological reports 11 , 12 , 13 , 14 . Recently, a case of Down syndrome was genetically identified in Neolithic Ireland (3629–3371 BCE), although no physical description was given 15 . However, we are aware of no prehistoric or historic cases of either trisomy 13 or 18 that have been identified genetically or osteologically.
The lack of genetically identified historic and prehistoric cases of autosomal trisomies is likely due to two factors. First, the rates of prevalence of Down, Edwards and Patau syndrome today are 1:705, 1:3226 and 1:7143, respectively (when considering live births, stillborn and terminated pregnancies) 9 and, assuming similar population demographics, it can be expected that these genetic disorders occurred at the same rates in past human populations 14 . Hence, probabilistically, researchers could not have reasonably expected to find cases until ancient DNA (aDNA) data sets became large enough. Second, methods for analysing copy number variations for modern data require long read lengths, relatively high depth of coverage and read-pair information, which are unlikely when working with aDNA 16 , 17 . Instead, we focus on the probabilities of reads mapping to whole chromosomes, allowing aDNA to be analysed at extremely low depths of coverage. Hence, to date, this is the first systematic genetic screening and osteological description of such cases in premodern samples.
We identify six cases of Down syndrome and one case of Edwards syndrome in a survey of 9,855 prehistoric and historic human genomes from across the globe, which are diagnosed using a novel Bayesian method designed for use with aDNA. This method can be applied to even low-coverage shotgun screened data, requiring as few as 1000 DNA sequence reads that have been mapped to the human genome. As no single osteological marker is pathognomonic for these genetic disorders, none of these cases was confirmed osteologically. However, when skeletal preservation and completeness was sufficient, we record all observed pathological lesions, and match these to osteological markers which are consistent with a diagnosis of the trisomy. By integrating evidence from archaeological contexts, we then describe the care with which many of these individuals were buried. Although it is unclear whether these individuals were identified as different by their communities of care, there is clearly no evidence that they were stigmatised by their communities in the past.
Genetic results
We screen shotgun sequencing data from 9855 prehistoric and historic individuals (see “Methods” for descriptions of laboratory protocols, quality control measures and data processing methodology). For each individual, we record the number and proportion of reads that mapped to each of the autosomal chromosomes.
We identify six new positive cases of Down syndrome, two genetically female and four genetically male. As expected, the positive cases for Down syndrome showed an approximately 1.5-fold higher proportion of reads mapping to chromosome 21 (range: 1.44–1.52) when compared to the mean proportion for negative cases (Fig. 1 ). All normalised posterior probabilities are one (to machine precision) for each positive case.

Filled black circles show individuals for which there were at least 1000 reads, and for which at most one chromosome was significantly overrepresented. Labels indicate individual IDs. Source data is provided as a Source data file.
We also reanalyse and confirm a previously published case of trisomy 21, the infant PN07 who was interred in a large Neolithic portal tomb in Poulnabrone, Ireland (3629–3371 BCE) 15 , showing that our method can be applied to sequencing data produced in any laboratory.
Among the 9855 screened individuals, we also identify one case (CRU013) of the comparatively rarer Edwards syndrome. The proportion of reads that mapped to chromosome 18 is 1.47 times greater than for the median for the negative group (Fig. 1 ) and the normalised posterior probability of the individual carrying Edwards syndrome is one (to machine precision). The individual was found to be genetically female, in line with the fact that Edwards syndrome is observed in genetically female individuals more often than in genetically male individuals by a ratio of 3:2 18 .
Archaeological contexts
Of the six newly identified cases of Down syndrome, five date to between 5000 and 2400 years ago and were intramural burials (burials within settlements and not in dedicated necropoleis). The remaining case dates to the eighteenth century CE and was found within a Finnish church cemetery. We present these results in chronological order (Table 1 ).
The earliest case in our new data, YUN039 (2898–2700 cal BCE), is a 6-month-old, genetically female individual, discovered in a ceramic-vessel buried under the floor of a dwelling from the Early Bronze Age layer of Tell Yunatsite, from the Pazardzhik Province of southern Bulgaria. YUN039 was not found with any burial items 19 , 20 . LAZ019 (1398–1221 cal BCE) is a 12–16-month-old, genetically female individual, discovered buried in a tiny cist grave in a yard-like area within the confines of a dwelling at the Mycenaean site of Lazarides on the island of Aegina, Greece 21 . LAZ019 was buried wearing a necklace made of 93 beads, glass paste, faience and carnelian (4 beads), of different colours and sizes 22 .
CRU001 (600-400 BCE), CRU024 (779-549 BCE) and ERE004 (801-764 BCE) were neonatal individuals, aged ~38, ~28 and ~26 weeks gestation at the time of death, respectively 23 , 24 , 25 , 26 , 27 . CRU001 and ERE004 are genetically male, and CRU024 is genetically female. These remains were excavated from burials beneath dwellings found at the site of Alto de la Cruz (CRU) and Las Eretas (ERE), two of the first proto-urban centres in Early Iron Age Navarra, Spain. CRU024 was buried with rich grave goods, including bronze rings, a Mediterranean seashell, and surrounded by the complete remains of three sheep and/or goats. While CRU001 and ERE004 were apparently buried in common dwellings, CRU024 was found buried in a structure with a large, decorated fireplace, which was likely a place of ritual.
Also found at the site of Alto de la Cruz in an intramural burial was individual CRU013 (600-400 BCE), a positive case of Edwards syndrome, who was estimated to have died at ~40 weeks of gestation.
Finally, HKI002 (1640–1790 CE) was found buried in a wooden coffin under present-day Senate Square in Helsinki, previously a church graveyard in Post-Medieval Finland. The funeral attire of HKI002 contained bronze pins and decorative bronze flowers, which were a common trend of the time.
Rates of prevalence
The six observed cases of Down syndrome in our data indicate a past rate of prevalence of ~1:1643, which is significantly lower than the overall modern rate of prevalence for Down syndrome of 1:705 9 . It is known that the age of the mother is strongly associated with an increased rate of prevalence 9 . When compared to the rate of prevalence for present-day mothers under the age of 20 (1:1282), we continue to find a lower rate of prevalence in past populations (Fig. 2 ), although this difference is not statistically significant. However, past rates of prevalence could be underestimated as infant burials are generally under-represented in the archaeological record 28 . Similarly, it is possible that the average age of mothers was lower due to an increased risk of death for the mother during childbirth in pre-modern times and assisted reproductive technologies allowing increased instances of motherhood at older ages in modern populations, although this lower mean age may be offset by an increased age of menarche in premodern populations 9 , 29 , 30 , 31 , 32 .

The red dashed line indicates the total rate of prevalence, ignoring mother’s age, and the blue dashed line indicates the observed rate of prevalence in the premodern individuals. Source data is provided as a source data file.
The single case of Edwards syndrome indicates a premodern rate of prevalence of 1:9855, which is not statistically significantly different from the modern prevalence of 1:3226 ( p ≈ 1) 9 . Finally, observing no cases of Patau syndrome in 9855 individuals is not unexpected, given that the modern rate of prevalence is 1:7143 ( p ≈ 1) 9 . However, we caution that Edwards and Patau syndrome almost always result in pre- or perinatal deaths. Given that perinatal remains will often be poorly preserved due to reasons such as reduced bone mineralisation and smaller bone size, thus making their bones more vulnerable to destruction and their discovery more difficult, our rates of prevalence are likely underestimated 33 .
Osteological results—trisomy 21
In this section, we identify osteological markers in our sample of seven individuals that could be indications of autosomal trisomies. We report all skeletal pathologies that are either known to be related to Down or Edwards syndrome or that are observed in more than one individual. However, we caution that osteological analyses alone are insufficient for such diagnoses, as no osteological symptom is pathognomonic for trisomy 21 or 18. Moreover, a large proportion of these features could be linked to environmental factors, such as malnutrition, and to other genetic disorders that can result in abnormal growth patterns or could even fit into normal growth variability 2 . However, the genetic diagnosis of these individuals offers an opportunity to suggest a relationship of the observed features to the identified chromosomal abnormality, directly or as a consequence. The osteological study of the individuals discussed in the present work are further complicated for two reasons: first, the skeletons are incomplete and often poorly preserved (see Supplementary Methods). Second, our cases are either late fetal losses, stillbirths or, at most, 16 months of age, and not all markers may be observable 2 .
To date, studies evaluating the skeletal manifestations of individuals with Down syndrome in past populations are few in number and often too statistically underpowered to answer questions about skeletal development, although studies based on modern individuals have shown that porosity and skeletal health issues, shortened femur length and reduced bone mineral density, are significantly more prevalent in cases of Down syndrome 34 . Several skeletal abnormalities have been identified in fetuses with Down syndrome, including: short stature with reduced length of long bones, microcephaly, brachycephaly, absence of nasal bone ossification and hypoplasia of the middle phalanx of the fifth digit 35 . Among the skeletal manifestations of Down syndrome that develop later in childhood are scoliosis, pes planus and Perthes disease 35 . Hence, we were motivated to look for abnormalities in the skeletons of the individuals that were genetically diagnosed as carrying trisomy 21.
Hand phalanges ossify at around 24 weeks of gestation, but they are so small that they are rarely recognised or correctly identified during excavation, thus making systematic recording impossible. Vitamin C, D, A, complex B and/or iron deficiency has also been associated with Down syndrome, individually or in combination, likely arising as a secondary complication of features of Down syndrome such as feeding difficulties 36 . Specifically, these deficiencies are commonly associated with cranial porosities including locations such as the sphenoid body, the external cranial vault, the superior aspect of the eye orbit (or cribra orbitalia ), as well as on long bones, which can be anticipated in individuals with trisomy 21 37 .
Despite the different stages of completeness and preservation throughout the cases, there is overlap in the anthropological observations, with the most striking being the reported growth disorders on bones, followed by porosities in the cranial elements. Overall, we observe porosity in at least one of the bones of the cranium in 4/6 cases (Supplementary Figs. 2 and 3 ), visible in the orbital roofs (2/3), mandible (3/4), the upper palate (2/3), the frontal and parietal bones (3/4), and/or the occipital squama (4/6). We also observe additional bone formation on the jugular limb of the pars lateralis in both cases for which the occipital bone was sufficiently well preserved for observation. Less common were visible striae or pitting of either the long bones (1/3) or the ilium (1/2). HKI002 was the only individual for which we observed (Supplementary Fig. 4 ) inner bone (incus) malformation (1/1), and dental enamel hypoplasia 38 (1/1) (Supplementary Fig. 5 ).
Other traits that can be associated with trisomy 21 overlap in three instances: individuals CRU001 and HKI002 present irregular bone growth on the pars lateralis segment of the occipital bone (e.g., flange, Supplementary Fig. 6 ), LAZ019 and YUN039 present morphological changes on the femora (Supplementary Fig. 5 ) (flattened and widened distal metaphysis, and antero-posterior flexion of the femur diaphysis, respectively) as well as cribra orbitalia and porotic hyperostosis . Additionally, HKI002 and YUN039 present signs of Vitamin C deficiency which often co-occurs with trisomy 21. CRU001 also displays a protrusion of the occipital squama, a cranial trait associated with trisomy 21 39 (Supplementary Fig. 7 ). A compilation of the traits recorded for each individual can be seen in Supplementary Table 1 , and we stress that some of these observed markers, such as dental hypoplasia and bone porosity, can be caused by many other diseases and even normal growth processes.
Osteological results—trisomy 18
Edwards syndrome is associated with very severe physical symptoms 40 , 41 , although very few clinical studies describe skeletal malformation 42 . In total, CRU013 presents numerous bone anomalies, some not being previously reported in the literature, such as: an abnormally shaped left scapula (Supplementary Figs. 8 and 9 ) which could be linked to describe chest malformations 43 , irregular morphology of the left hemiarch of the axis 40 (Supplementary Fig. 10 ), extremely thin humeral diaphysis and pronounced curvature of the epiphyseal surfaces for both humeri (Supplementary Fig. 11 ), thinning of the diaphysis of the right femur (left missing) and the left tibia (right missing) (Supplementary Fig. 12 ). Furthermore, an incomplete bone fragment, likely the diaphysis of a fibula and not a clavicula, shows an unusual twist (Supplementary Fig. 13 ). The age of CRU013 was estimated at 40 weeks gestation when using the maximal length of both humeri (Supplementary Table 2 ). Of note, however, is the underdevelopment of the distal width of the right humerus, according to which the age estimate for the same individual would have been only 30 weeks of gestation. We thus caution that estimating the age of the individual using long bones, when these bones are shown to have undergone irregular development, and when short stature is a common symptom of trisomy 18, will likely produce downward-biased estimates.
In this analysis of 9855 prehistoric and historic individuals, we detect six cases of Down syndrome, and one case of Edwards syndrome. In addition, we confirm one published case of Down syndrome. These individuals, all of whom died either before or shortly after birth, or at most 16 months of age, come from Neolithic Ireland (~3500 BCE), Bronze Age Bulgaria (~2700 BCE) and Greece (~1300 BCE), Iron Age Spain (~600 BCE), or Post-Medieval Finland (~1720 CE).
For the individuals who we identify as having Down syndrome, we observe evidence of porosity in the cranial bones for almost all individuals, and abnormal growths on the ossis occipitalis and pars lateralis in both cases where these bones were sufficiently preserved. We also observe additional evidence of abnormal bone growth in occipital protrusions, inner ear bone malformation and enamel hypoplasia. Abnormal bone development, such as porosity, has been shown to occur in individuals with Down syndrome as they age, and it has also been observed in some elements in fetal development. Studies on a mouse model for Down syndrome (Ts65Dn) indicate a disruption of bone development and homoeostasis caused by trisomies of genes associated with human bone development, even as early as during the development of the fetus 44 , 45 . However, we stress that skeletal porosity can be caused by a number of factors, such as: taphonomic factors, many diseases of different cause, the physiological growth at different ages of the individual and the mother’s health 46 , 47 .
The skeleton associated with the case of Edwards syndrome presents with severe skeletal abnormalities, some of them being consistent with the genetic diagnosis of trisomy 18. Hyperostosis and morphological abnormalities were observed in almost all of the preserved bones. The age-at-death for this individual was estimated at 40 weeks gestation. Based on osteological criteria it is impossible to determine if this individual was stillborn or born alive.
In all reported cases, the burials of the individuals were either special, or performed with care according to standard practices. PN07 was buried in a large portal tomb in Neolithic Ireland. All of the Bronze Age or Iron Age burials were intramural burials, indicating that these infants were considered worthy of deserving a burial place inside of the dwellings. Finally, HKI002 was afforded a normal Christian burial, with burial dress following the trend of the period.
LAZ019 (12–16 months) and YUN039 (6 months) are the only cases which definitely survived postnatally. Down syndrome is associated with more than skeletal symptoms 48 , 49 . Affected children may have additional disorders, such as congenital heart defects, lung diseases, neurodevelopmental disorders, hearing loss, vitamin and nutrient deficiencies and possible accompanying infections. Hence, newborns with Down syndrome usually require additional care. The choice to devote more time to these individuals, suggests parents, caretakers and possibly a community with compassion that would not stigmatise or ignore those in need 50 , 51 , 52 . Though signs of a head injury have been recorded for individual YUN039 (i.e. extensive haemorrhages), the cause may not be interpersonal violence. The prevalence of epilepsy in Down syndrome is up to 26% and an epileptic seizure could be one of very many potential explanations for head injury in a child with Down syndrome 53 . Additionally, the haemorrhages could also be consistent with scurvy or inflammatory processes 37 . Overall, mortuary treatment provides a good indication of the attitudes communities had towards these individuals. All examples described in this study were cared for after death through various rituals, which show, in some cases exceptional, recognition of them as community members.
Although the frequency of Down syndrome cases among our tested individuals is significantly lower than the present-day rate of prevalence at birth, we caution that this difference could be explained by non-random sampling and differential sample preservation. Specifically, the remains of stillbirths, infants and children are less likely to be preserved, and are buried in prehistoric and ancient cemeteries less often than adults 28 , 33 , 54 , 55 , 56 Additionally, since all cases came from either stillbirths or infants, the skeletons are more fragile, less likely to be preserved and often incomplete 33 . Despite observing a lower frequency of cases of Down Syndrome, we find a total of three individuals with Down syndrome at two geographically close Iron Age sites in Spain. Additionally, the sole case of Edwards syndrome was also discovered at one of the sites. It should be noted that while the dominant burial custom in this region at the time was cremation, some perinates and infants received intramural burials. However, when considering the number of infants that have been discovered and sequenced at these specific sites thus far, the number of cases of trisomies is surprisingly high 57 . Further research will be needed to corroborate the high frequency and to form hypotheses on the cultural practices that may have led to this.
Skeletal manifestations alone cannot be used as predictors for the presence of Down syndrome or Edwards syndrome, especially in archaeological assemblages that lack complete crania or complete long bones, such as is common in infants 33 . The manifestations of the associated traits of these syndromes are variable, and the presented cases here display levels of overlapping osteological markers, especially in the growth disorders, which feature as the primary observation in all cases. Porosities of the cranial elements follow closely in occurrence, highlighting the already established suggestion that diagnosis of such lesions be treated with caution, since they could be an expression of multiple and/or more complex pathologies.
As the aDNA record continues to grow, genetic disorders with extremely low rates of prevalence will be able to be more frequently discovered. Methods for screening data, such as the one presented here, will need to be developed and refined for better detection of these afflictionsis. Integrated with contextual and anthropological data, they afford a perspective into the way that these disorders were viewed and treated in past communities. All individuals that we identified as carrying trisomy 18 and 21 may not have been visually identifiable as “different” by their communities, given that they died in infancy or earlier. We find it notable that individuals with trisomy 18 and 21 were buried according to standard practices, or in a few cases, given exceptional burials or elaborate grave goods.
Note added in proof : A recent publication by Anastasiadou et al. 58 identified an infant with trisomy 21 from Iron Age Britain using ancient DNA analysis.
All samples were collected for their original studies with the appropriate permits as required by the laws in their associated regions. We confirm that we followed established ethical guidelines for archaeogenetic research.
DNA sequencing and analysis
In this study, we analyse Illumina shotgun sequencing data collected from 2016 to 2022 at the Department for Archeaogenetics at the Max Planck Institute for Evolutionary Anthropology (formerly at the Max Planck Institute for the Science of Human History). Most samples are sourced from survey sequencing (used to determine sample quality for deeper downstream sequencing) of around 5–10 million reads. After adaptor trimming, reads were aligned to the human reference genome GRCh37 using the Burrows-Wheeler Aligner. Sequence reads were counted for all autosomal chromosomes after filtering for a minimum sequence length of 35 and a mapping quality of at least 25.
Estimating posterior probabilities of cases of trisomies and calculating Z -scores
To screen the full data set for trisomy, we assume a binomial distribution for the proportion of reads that map to chromosome \(c\in \{1,..,22\}\) . To account for overdispersion caused by different DNA sequencing library protocols, sequencing runs and artefacts due to variable DNA preservation, we also assume a prior beta distribution for the binomial probability parameter governing the probability of mapping a read to chromosome \(c\) , denoted \({p}_{c}\) . We estimate the parameters of the beta from a filtered subset of the data, using only samples with at least 10,000 reads (7103 samples were retained based on this criterion). Additionally, we also estimate these distributions independently for each observed type of library protocol. Note that this cut-off for parameter estimation is different to the cut-off of 1000 reads for downstream screening.
The Z -score for the observed proportion of reads ( \({\hat{p}}_{i,c}\) ) mapping to chromosome \(c\) for individual \(i\) , denoted \({Z}_{i,c}\) , is calculated as the number of standard deviations of the observed proportion of reads mapping to chromosome \(c\) , compared to expectation via a beta-binomial distribution, as plotted in Supplementary Fig. 1 . For a full derivation of the formulae, see Supplementary Methods Section 1 .
Statistical testing
We estimate the beta-binomial parameters using a maximum-likelihood approach via the beta.mle() function in the Rfast (v2.0.6) package 59 . Probability density calculations for the beta-binomial distribution are calculated using the dbbinom() function in the extraDist (v1.9.1) package 60 . Tests comparing the observed rates of prevalence to the expected modern rates of prevalence are performed using the binom.test() function in the stats package. All statistical plots are created using the ggplot2 package 61 .
Genetic sex estimates
To estimate the genetic sex, we compare the overall fraction of reads mapping to the X and Y chromosomes for each individual, denoted \({p}_{i,x}\) and \({p}_{i,y}\) , as in Roca-Rada et al. 62 . Individuals that fell within the cluster with the mean, indicating no Y chromosome, are assigned as genetically female, and the remaining individuals are assigned as genetically male.
Osteological analyses
No reanalysis of the skeletal remains were performed. All results are collected from a reanalysis of the original publications, photographs and osteological reports. The age-at-death of infants who died close to or before birth cannot be confidently estimated until a certain stage of osteological development is reached 2 . Hence, we use multiple skeleton age estimators, based on different proposals made by researchers from forensic medicine and obstetrics 63 , 64 , 65 , 66 , 67 , 68 . In the case of perinatal individuals, we could only use the length of the preserved long bones to obtain an approximate age.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
No new DNA data were generated for this study. The sequence data has been processed and we provide the proportions of reads mapping to the autosomes and total read counts as a tsv at (github.com/BenRohrlach/TrisomyAncientDNAStudy 69 ). However, sample names and sequencing protocols for non-reported individuals have been anonymised. The read count, and source data for Figs. 1 and 2 and Supplementary Fig. 1 can be found as Supplementary Data and at github.com/BenRohrlach/TrisomyAncientDNAStudy 69 . The current location of the remains that are analysed in this study (CRU001, CRU013, CRU024, ERE004, HKI002, LAZ019 and YUN039) are described in the Supplementary Methods, and the DNA sequences reported in this paper have been deposited in the European Nucleotide Archive under the accession number PRJEB71003 . Source data are provided with this paper.
Acknowledgements
The authors wish to thank Professor Nigel Bean, Dr Elina Salmela, Dr Vincent Braunack-Mayer, Associate Professor Denice Higgins, Dr Jaana Hurnanen, Dr Olli Varkkola and Heini Hämäläinen (Helsinki City Museum) for enlightening and instructive conversations. This study was supported by the Max Planck Society and the European Research Council (ERC) under the European Union’s Horizon 2020 Research and Innovation Program Grant 771234-PALEoRIDER (to W.H.) and 851511 (to S.S.). J.C.T. is supported by the Australian Research Council (ARC) through a Discovery Early Career Researcher Award (DE210101235). The Max Planck - Harvard Research Center for the Archaeoscience of the Ancient Mediterranean (MHAAM) and 101001951-MySocialBeIng (to P.W.S.). R.R. is supported by The Catalan Institution for Research and Advanced Studies of the Generalitat de Catalunya.
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and affiliations.
Department of Archaeogenetics, Max Planck Institute for Evolutionary Anthropology, Leipzig, Germany
Adam Benjamin Rohrlach, Maïté Rivollat, Anthi Tiliakou, Angela Mötsch, Philipp W. Stockhammer, Stephan Schiffels, Johannes Krause, Wolfgang Haak & Kay Prüfer
School of Computer and Mathematical Sciences, University of Adelaide, Adelaide, SA, Australia
Adam Benjamin Rohrlach & Jonathan Tuke
ArcheOs lab, Department of Archaeology, Ghent University, Sint-Pietersnieuwstraat 35, 9000, Gent, Belgium
Maïté Rivollat
Archaeo-DNA lab, Department of Archaeology, Durham University, Lower Mount Joy, South Road, Durham, DH1 3LE, UK
De la Préhistoire à l’Actuel, Culture, Environnement, Anthropologie - UMR 5199, Bordeaux University, Bât. B8, Allée Geoffroy Saint Hilaire, CS50023, 33615, Pessac cedex, France
Department of Prehistory, Archaeology, Ancient History and Greek and Latin Philology, INAPH, University of Alicante, San Vicente del Raspeig, Spain
Patxuka de-Miguel-Ibáñez
Sociedad de Ciencias Aranzadi, Donosti, Spain
Hospital Verge dels Lliris, Alcoi, Alicante, Spain
Department of Biology, University of Turku, Turku, Finland
Ulla Moilanen & Päivi Onkamo
Department of Archaeology, University of Turku, Turku, Finland
Anne-Mari Liira
Evolution of Cultural Diversity Initiative, Australian National University, Canberra, ACT, Australia
João C. Teixeira
Australian Centre for Ancient DNA, School of Biological Sciences, University of Adelaide, Adelaide, SA, Australia
João C. Teixeira, Xavier Roca-Rada & Bastien Llamas
Centre of Excellence for Australian Biodiversity and Heritage, University of Adelaide, Adelaide, SA, Australia
João C. Teixeira & Bastien Llamas
CEIS.20 Centro de Estudos Interdisciplinares, Universidade de Coimbra, Coimbra, Portugal
Departamento de Ciencias Humanas y de la Educación, Universidad Pública de Navarra, Pamplona, Spain
Javier Armendáriz-Martija
National Archaeological Institute with Museum at the Bulgarian Academy of Sciences, Saborna str. 2, Sofia, Bulgaria
Kamen Boyadzhiev & Yavor Boyadzhiev
National Centre for Indigenous Genomics, Australian National University, Canberra, ACT, Australia
Bastien Llamas
Telethon Kids Institute, Indigenous Genomics Research Group, Adelaide, SA, Australia
Max Planck-Harvard Research Center for the Archaeoscience of the Ancient Mediterranean (MHAAM), Max Planck Institute for Evolutionary Anthropology, Deutscher Platz 6, Leipzig, Germany
Angela Mötsch & Philipp W. Stockhammer
Hellenic Center for Bioarchaeology, Athens, Greece
Eleni-Anna Prevedorou
Department of History and Archaeology, National and Kapodistrian University of Athens, Athens, Greece
Naya Polychronakou-Sgouritsa
Department of Anthropology, Arizona State University, Tempe, AZ, USA
Jane Buikstra
Department of Biosciences, University of Helsinki, Helsinki, Finland
Päivi Onkamo
Institute for Pre- and Protohistoric Archaeology and Archaeology of the Roman Provinces, Ludwig Maximilian University, Geschwister-Scholl-Platz 1, München, Germany
Philipp W. Stockhammer
Hasso-Plattner-Institute, University of Potsdam, Potsdam, Germany
Henrike O. Heyne
Hasso Plattner Institute, Mount Sinai School of Medicine, New York, USA
Finnish Institute for Molecular Medicine (FIMM), University of Helsinki, Helsinki, Finland
Institute of Human Genetics, University of Leipzig Medical Center, Leipzig, Germany
Johannes R. Lemke
Center for Rare Diseases, University of Leipzig Medical Center, Leipzig, Germany
Departament de Prehistòria, Universitat Autònoma de Barcelona, Bellaterra, Spain
Roberto Risch
You can also search for this author in PubMed Google Scholar
Contributions
ABR and KP designed the study and SS, WH and JK edited the manuscript. ABR and KP collected and processed all of the data. ABR, KP, MR, PdMI, AT and RR wrote the first draft of the manuscript. ABR designed the Bayesian method, and ABR and KP performed all statistical analyses. JAM, KB, YB, PO, RR, UM, AML and PWS wrote the archaeological sections. All authors contributed to editing the final manuscript.
Corresponding authors
Correspondence to Adam Benjamin Rohrlach or Kay Prüfer .
Ethics declarations
Competing interests.
The authors declare no competing interests.
Peer review
Peer review information.
Nature Communications thanks Julia Gresky, Toomas Kivisild and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information, peer review file, reporting summary, source data, rights and permissions.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ .
Reprints and permissions
About this article
Cite this article.
Rohrlach, A.B., Rivollat, M., de-Miguel-Ibáñez, P. et al. Cases of trisomy 21 and trisomy 18 among historic and prehistoric individuals discovered from ancient DNA. Nat Commun 15 , 1294 (2024). https://doi.org/10.1038/s41467-024-45438-1
Download citation
Received : 02 April 2023
Accepted : 19 January 2024
Published : 20 February 2024
DOI : https://doi.org/10.1038/s41467-024-45438-1
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
By submitting a comment you agree to abide by our Terms and Community Guidelines . If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing: Translational Research newsletter — top stories in biotechnology, drug discovery and pharma.

IMAGES
VIDEO
COMMENTS
View Unfriended Case Study Student Guide.pdf from ART 1000 at Seminole State College of Florida. UN-FRIENDED DNA CASE STUDY Directions: As you watch the 20/20 Investigation, answer the. AI Homework Help. ... Please refer to the attachment to answer this question. This question was created from BUS 1700 Course Project - Grading Worksheet.xlsx. ...
View Unfriended Case Study Student Guide.pdf from FIN 1 at Mather High School. UN-FRIENDED DNA CASE STUDY Directions: As you watch the 20/20 Investigation, answer the following questions. 1. What. AI Homework Help. Expert Help. Study Resources. ... View Un-Friended DNA Case Study (1).pdf from CRJU 2150 at Georgia Piedmont Technical Co...
Study with Quizlet and memorize flashcards containing terms like Why was the case of Colin Pitchfork important in forensics? (there are multiple answers), Who was the first criminal in the United States to be prosecuted using DNA evidence?, What was the importance of heteroplasmy in this case? How did it aid in identifying the remains? and more.
View Kami Export - Josue Santiago - Unfriended Case Study Student Guide (1).pdf from SCIENCE 542 at Trenton Central High. UN-FRIENDED DNA CASE STUDY Directions: As you watch the 20/20 Investigation,
1. Uploaded by SuperHumanMantisPerson1090 on coursehero.com. Helpful. Unhelpful. Home /. Psychology. UN - FRIENDED DNA CASE STUDY Directions: As you watch the 20/20 Investigation, answer the following questions. 1. What evidence led the parents to believe that Skylar willingly left (snuck out of) her house?
He and Christakis looked at almost 2,000 participants and identified about 1,400 pairs of friends. The findings, published this week in the Proceedings of the National Academy of Sciences, show ...
UN-FRIENDED DNA CASE STUDY Directions: As you watch the 20/20 Investigation, answer the following questions. 1. What evidence led the parents to believe that Skylar willingly left (snuck out of) her house? the video from the surveillance showed her getting in the car 2. The friends say that when th ... Purchase document to see full attachment
25. True or False: Fraternal twins have identical DNA. false. Complete this sentence: "Lillian... ." Select all that apply. -is the biological mother of her children. -is her own fraternal twin. -has 2 distinct sets of DNA. -is genetically female.
Pooling resources, as well as collaborating with professionals outside of forensic anthropology, is a useful strategy to pursue when anthropological methods are exhausted.The case study presented here demonstrates a collaborative approach between forensic anthropologists, forensic genetic genealogists, and law enforcement in a century-old homicide.
Study with Quizlet and memorize flashcards containing terms like PART 1: Jason, a 12-year-old boy, has been sick for the last 3 days with flulike symptoms: pallor, fatigue, loss of appetite, muscle aches, and a 101° F to 102° F fever. His parents have been giving him over-the-counter cough and cold medications as well as Tylenol to control his fever. His parents take him to his pediatrician ...
View Un-Friended DNA Case Study (1).pdf from CRJU 2150 at Georgia Piedmont Technical College. ... at Georgia Piedmont Technical College. UN-FRIENDED DNA CASE STUDY Directions: As you watch the 20/20 Investigation, answer the following. AI Homework Help. Expert Help. Study Resources. ... Kami Export - Josue Santiago - Unfriended Case Study ...
The video case study is presented in eight modules, each followed by embedded questions. Educators can use the extension questions listed below for additional class discussion or written assessments. Modules 1 and 2 establish the genetic basis of RP and describe other features of the disease. Prior to starting the video, prompt students by ...
In the answer sequence above and in the rest of the answer key, the variant nucleotide change is represented in . red. text and the additional nucleotide change is represented in . blue . text. 2. What gene are the scientists looking at? SLC52A2 (Solute Carrier Family 52 Member 2) 3.
1. The family tree shows that Melissa Smith and Robert Smith are third cousins to Inez Johnson and George Underhill. Both Melissa and Robert share DNA with Inez and George at an expected level. According to the 2016 data from the Shared cM Project, 3C has an average of 79 cM shared with a range from 0 to 200 cM or so.
This page titled 8.1: Case Study: Genes and Inheritance is shared under a CK-12 license and was authored, remixed, and/or curated by Suzanne Wakim & Mandeep Grewal via source content that was edited to the style and standards of the LibreTexts platform; a detailed edit history is available upon request. People tend to look similar to their ...
This comprehensive module explores the complex role of DNA in a sexual assault investigation, including the alternative sources of DNA evidence and their potential significance or impact on a sexual assault investigation. A number of resources and tools are provided, along with a series of complex and interactive case examples.
FORENSIC SCIENCE 101. MateKingfisher4017. 2/27/2024. View full document. UN- FRIENDED DNA CASE STUDY Directions: As you watch the 20/20 Investigation, answer the following questions. 1. What evidence led the parents to believe that Skylar willingly left (snuck out of) her house? The screen on her window was gone and the window was slightly open.
Figure 5.1.1 Family tree - three generations. People tend to carry similar traits to their biological parents, as illustrated by the family tree. Beyond just appearance, you can also inherit traits from your parents that you can't see. Rebecca becomes very aware of this fact when she visits her new doctor for a physical exam.
Interestingly, three cases of trisomy 21, and the case of trisomy 18 were detected in two contemporaneous sites in early Iron Age Spain (800-400 BCE), potentially suggesting a higher frequency of ...
Extend. Students will select a level 1 and a level 2 genetic disorder. They will research the disorders and prepare an informational poster for a walk-about. Evaluate. Students will use academic vocabulary cards to demonstrate conceptual relationships and their understanding of basic DNA processes.
Using the coding techniques described in this chapter, carefully read through the case study and determine the most accurate CPT code(s) and HCPCS code(s) and modifier(s), if appropriate. Answer Q&A
Putting the Pieces Together: The Discovery of DNA Structure and Replication. In the autumn of 1952, while the weather in London, England, was cooling down, the race to discover the structure of DNA was heating up inside two rival laboratories: the Cavendish Lab at Cambridge University and the laboratory of J.T. Randall at nearby King's College.