The age-by-disease interaction hypothesis of late-life depression
Affiliation.
- 1 Department of Psychiatry, University of Pittsburgh, Pittsburgh, PA 15219, USA.
- PMID: 23570886
- PMCID: PMC3549303
- DOI: 10.1016/j.jagp.2013.01.053
The phenomenologic diagnosis of depression is successful in increasing diagnostic reliability, but it is a classification scheme without biologic bases. One subtype of depression for which evidence suggests a unique biologic basis is late-life depression (LLD), with first onset of symptoms after the age of 65. LLD is common and poses a significant burden on affected individuals, caretakers, and society. The pathophysiology of LLD includes disruptions of the neural network underlying mood, which can be conceptualized as the result of dysfunction in multiple underlying biologic processes. Here, we briefly review current LLD hypotheses and then describe the characteristics of molecular brain aging and their overlap with disease processes. Furthermore, we propose a new hypothesis for LLD, the age-by-disease interaction hypothesis, which posits that the clinical presentation of LLD is the integrated output of specific biologic processes that are pushed in LLD-promoting directions by changes in gene expression naturally occurring in the brain during aging. Hence, the brain is led to a physiological state that is more susceptible to LLD, because additional pushes by genetic, environmental, and biochemical factors may now be sufficient to generate dysfunctional states that produce depressive symptoms. We put our propositions together into a decanalization model to aid in illustrating how age-related biologic changes of the brain can shift the repertoire of available functional states in a prodepression direction, and how additional factors can readily lead the system into distinct and stable maladaptive phenotypes, including LLD. This model brings together basic research on neuropsychiatric and neurodegenerative diseases more closely with the investigation of normal aging. Specifically, identifying biologic processes affected during normal aging may inform the development of new interventions for the prevention and treatment of LLD.
Copyright © 2013 American Association for Geriatric Psychiatry. Published by Elsevier Inc. All rights reserved.

Publication types
- Research Support, N.I.H., Extramural
- Aging / genetics*
- Aging / physiology*
- Brain / physiopathology*
- Cost of Illness
- Depression / diagnosis
- Depression / genetics*
- Depression / physiopathology*
- Genetic Predisposition to Disease / genetics*
- Homeostasis / physiology
- Neural Pathways / physiopathology
Grants and funding
- R01 MH093723/MH/NIMH NIH HHS/United States
- R01 MH077159/MH/NIMH NIH HHS/United States
- MH093723/MH/NIMH NIH HHS/United States
- K02 MH084060/MH/NIMH NIH HHS/United States
- UL1 TR000005/TR/NCATS NIH HHS/United States
- MH084060/MH/NIMH NIH HHS/United States
Advertisement
The inflammatory & neurodegenerative (I&ND) hypothesis of depression: leads for future research and new drug developments in depression
- Original Paper
- Published: 16 December 2008
- Volume 24 , pages 27–53, ( 2009 )
Cite this article
- Michael Maes 1 ,
- Raz Yirmyia 2 ,
- Jens Noraberg 3 , 4 ,
- Stefan Brene 5 ,
- Joe Hibbeln 6 ,
- Giulia Perini 7 ,
- Marta Kubera 8 ,
- Petr Bob 9 ,
- Bernard Lerer 10 &
- Mario Maj 11
7715 Accesses
675 Citations
10 Altmetric
Explore all metrics
Despite extensive research, the current theories on serotonergic dysfunctions and cortisol hypersecretion do not provide sufficient explanations for the nature of depression. Rational treatments aimed at causal factors of depression are not available yet. With the currently available antidepressant drugs, which mainly target serotonin, less than two thirds of depressed patients achieve remission. There is now evidence that inflammatory and neurodegenerative (I&ND) processes play an important role in depression and that enhanced neurodegeneration in depression may–at least partly–be caused by inflammatory processes. Multiple inflammatory-cytokines, oxygen radical damage, tryptophan catabolites–and neurodegenerative biomarkers have been established in patients with depression and these findings are corroborated by animal models of depression. A number of vulnerability factors may predispose towards depression by enhancing inflammatory reactions, e.g. lower peptidase activities (dipeptidyl-peptidase IV, DPP IV), lower omega-3 polyunsaturated levels and an increased gut permeability (leaky gut). The cytokine hypothesis considers that external, e.g. psychosocial stressors, and internal stressors, e.g. organic inflammatory disorders or conditions, such as the postpartum period, may trigger depression via inflammatory processes. Most if not all antidepressants have specific anti-inflammatory effects, while restoration of decreased neurogenesis, which may be induced by inflammatory processes, may be related to the therapeutic efficacy of antidepressant treatments. Future research to disentangle the complex etiology of depression calls for a powerful paradigm shift, i.e. by means of a high throughput-high quality screening, including functional genetics and genotyping microarrays; established and novel animal and ex vivo–in vitro models for depression, such as new transgenic mouse models and endophenotype-based animal models, specific cell lines, in vivo and ex vivo electroporation, and organotypic brain slice culture models. This screening will allow to: 1) discover new I&ND biomarkers, both at the level of gene expression and the phenotype; and elucidate the underlying molecular I&ND pathways causing depression; and 2) identify new therapeutic targets in the I&ND pathways; develop new anti-I&ND drugs for these targets; select existing anti-I&ND drugs or substances that could augment the efficacy of antidepressants; and predict therapeutic response by genetic I&ND profiles.
This is a preview of subscription content, log in via an institution to check access.
Access this article
Price includes VAT (Russian Federation)
Instant access to the full article PDF.
Rent this article via DeepDyve
Institutional subscriptions
Similar content being viewed by others

Molecular Mechanisms of Psilocybin and Implications for the Treatment of Depression
Susan Ling, Felicia Ceban, … Roger S. McIntyre

Major Depressive Disorder: Advances in Neuroscience Research and Translational Applications
Zezhi Li, Meihua Ruan, … Yiru Fang

Animal models of depression: pros and cons
Jaanus Harro
Amodio P, De Toni EN, Cavalletto L, Mapelli D, Bernardinello E, Del Piccolo F, Bergamelli C, Costanzo R, Bergamaschi F, Poma SZ, Chemello L, Gatta A, Perini G (2005) Mood, cognition and EEG changes during interferon (αIFN) treatment for chronic hepatitis C. J. Affect. Disord. 84(1):93–98
PubMed CAS Google Scholar
Angelucci F, Brene S, Mathe AA (2005) BDNF in schizophrenia, depression and corresponding animal models. Mol. Psychiatry 10(4):345–352
Anisman H, Kokkinidis L, Merali Z (2002) Further evidence for the depressive effects of cytokines: anhedonia and neurochemical changes. Brain Beh. Immun. 16(5):544–556 Review
CAS Google Scholar
Anisman H, Merali Z, Poulter MO, Hayley S (2005) Cytokines as a precipitant of depressive illness: animal and human studies. Curr. Pharm. Design 11(8):963–972
Anisman H, Poulter MO, Gandhi R, Merali Z, Hayley S (2007) Interferon-alpha effects are exaggerated when administered on a psychosocial stressor backdrop: cytokine, corticosterone and brain monoamine variations. J. Neuroimmunol. 186(1–2):45–53
Anttila S, Huuhka K, Huuhka M, Rontu R, Hurme M, Leinonen E, Lehtimaki T (2007) Interaction between 5-HT1A and BDNF genotypes increases the risk of treatment-resistant depression. J. Neural. Transm. 114(8):1065–1068
Babcock TA, Carlin JM (2000) Transcriptional activation of indoleamine dioxygenase by interleukin 1 and tumor necrosis factor alpha in interferon-treated epithelial cells. Cytokine 12(6):588–594
Banerjee A, Jain G, Grover S, Singh J (2007) Mania associated with interferon. J. Postgraduate Med. 53(2):150
Bazan NG, Marcheselli VL, Cole-Edwards K (2005) Brain response to injury and neurodegeneration: endogenous neuroprotective signaling. Ann. NY Acad. Sci. 1053:137–147 Review
Beck RD Jr., Wasserfull C, Ha GK, Cushman JD, Huang Z, Atkinson MA, Petitto JM (2005) Changes in hippocampal IL-15, related cytokines, and neurogenesis in IL-2 deficient mice. Brain Res. 1041:223–230
Beltz BS, Sandeman DC (2003) Regulation of life-long neurogenesis in the decapod crustacean brain. Arth. Struct. Dev. 32:39–60
Google Scholar
Beltz BS, Tlusty MF, Benton JL, Sandeman DC (2007) Omega-3 fatty acids upregulate adult neurogenesis. Neurosci. Lett. 145(2):154–158
Benazzi F (2007a) Bipolar II Disorder : Epidemiology, Diagnosis and Management. CNS Drugs. 21(9):727–740
Benazzi F (2007b) Is there a continuity between bipolar and depressive disorders? Psychoth. Psychosom. 76(2):70–76
Berlim MT, Turecki G (2007) Definition, assessment, and staging of treatment-resistant refractory major depression: a review of current concepts and methods. Can. J. Psychiatry 52(1):46–54
PubMed Google Scholar
Bjornebekk A, Mathe AA, Brene S (2005) The antidepressant effect of running is associated with increased hippocampal cell proliferation. Int. J. Neuropsychopharmacol. 8(3):357–368
Bjørnebekk A, Mathé AA, Gruber SH, Brené S (2007) Social isolation increases number of newly proliferated cells in hippocampus in female flinders sensitive line rats. Hippocampus. 17(12):1193–1120
Bjørnebekk A, Mathé AA, Gruber SH, Brené S (2008) Housing conditions modulate escitalopram effects on antidepressive-like behaviour and brain neurochemistry. Int. J. Neuropsychopharmacol. 23:1–13
Bonaccorso S, Puzella A, Marino V, Pasquini M, Biondi M, Artini M, Almerighi C, Levrero M, Egyed B, Bosmans E, Meltzer HY, Maes M (2001) Immunotherapy with interferon-alpha in patients affected by chronic hepatitis C induces an intercorrelated stimulation of the cytokine network and an increase in depressive and anxiety symptoms. Psychiatry Res. 105(1–2):45–55
Bonaccorso S, Marino V, Biondi M, Grimaldi F, Ippoliti F, Maes M (2002a) Depression induced by treatment with interferon-alpha in patients affected by hepatitis C virus. J. Affect. Disord. 72(3):237–241
Bonaccorso S, Marino V, Puzella A, Pasquini M, Biondi M, Artini M, Almerighi C, Verkerk R, Meltzer H, Maes M (2002b) Increased depressive ratings in patients with hepatitis C receiving interferon-alpha-based immunotherapy are related to interferon-alpha-induced changes in the serotonergic system. J. Clin. Psychopharmacol. 22(1):86–90
Bremner JD, Narayan M (1998) The effects of stress on memory and the hippocampus throughout the life cycle: implications for childhood development and aging. Developm. Psychopathol. 10(4):871–885
Brown ES, Rush AJ, McEwen BS (1999) Hippocampal remodeling and damage by corticosteroids: implications for mood disorders. Neuropsychopharmacol. 21(4):474–484
Brown ES, J Woolston D, Frol A, Bobadilla L, Khan DA, Hanczyc M, Rush AJ, Fleckenstein J, Babcock E, Cullum CM (2004) Hippocampal volume, spectroscopy, cognition, and mood in patients receiving corticosteroid therapy. Biol. Psychiatry 55(5):538–545
Bukalo O, Fentrop N, Lee AY, Salmen B, Law JW, Wotjak CT, Schweizer M, Dityatev A, Schachner M (2004) Conditional ablation of the neural cell adhesion molecule reduces precision of spatial learning, long-term potentiation, and depression in the CA1 subfield of mouse hippocampus. J. Neurosci. 24:1565–1577
Campbell S, MacQueen G (2006) An update on regional brain volume differences associated with mood disorders. Curr. Opin. Psychiatry 19(1):25–33
Capuron L, Neurauter G, Musselman DL, Lawson DH, Nemeroff CB, Fuchs D, Miller AH (2003) Interferon-alpha-induced changes in tryptophan metabolism. relationship to depression and paroxetine treatment. Biol. Psychiatry 54(9):906–914
Carter CJ (2007) Multiple genes and factors associated with bipolar disorder converge on growth factor and stress activated kinase pathways controlling translation initiation: implications for oligodendrocyte viability. Neurochem. Int. 50(3):461–490
Castanon N, Bluthé RM, Dantzer R (2001) Chronic treatment with the atypical antidepressant tianeptine attenuates sickness behavior induced by peripheral but not central lipopolysaccharide and interleukin-1beta in the rat. Psychopharmacol. 154(1):50–60
Chavez AM, Menconi MJ, Hodin RA, Fink MP (1999) Cytokine-induced intestinal epithelial hyperpermeability: role of nitric oxide. Crit. Care Med. 27(10):2246–2251
Checa N, Canals JM, Alberch J (2000) Developmental regulation of BDNF and NT-3 expression by quinolinic acid in the striatum and its main connections. Exp. Neurology 165(1):118–124
Chung KK, Dawson TM, Dawson VL (2005) Nitric oxide, S-nitrosylation and neurodegeneration. Cell Mol. Biol. (Noisy-le-grand) 51(3):247–254 Review
Clark E, Hoare C, Tanianis-Hughes J, Carlson GL, Warhurst G (2005) Interferon gamma induces translocation of commensal Escherichia coli across gut epithelial cells via a lipid raft-mediated process. Gastroenterol. 128(5):1258–1267
Contestabile A (2001) Oxidative stress in neurodegeneration: mechanisms and therapeutic perspectives. Curr. Topics Med. Chemistry 1(6):553–568 Review
Coti Bertrand P, O'Kusky JR, Innis SM (2006) Maternal dietary (n-3) fatty acid deficiency alters neurogenesis in the embryonic rat brain. J. Nutr. 136(6):1570–1575
Cowen PJ (2002) Cortisol, serotonin and depression: all stressed out? Brit. J. Psychiatry 180:99–100
de Jonge WJ, van der Zanden EP, The FO, Bijlsma MF, van Westerloo DJ, Bennink RJ, Berthoud HR, Uematsu S, Akira S, van den Wijngaard RM, Boeckxstaens GE (2005) Stimulation of the vagus nerve attenuates macrophage activation by activating the Jak2-STAT3 signaling pathway. Nature Immunol 6(8):844–851
Dinan T (2001) Novel approaches to the treatment of depression by modulating the hypothalamic—pituitary—adrenal axis. Hum. Psychopharmacol. 16(1):89–93
Duman RS (2002) Pathophysiology of depression: the concept of synaptic plasticity. Eur. Psychiatry 17(Suppl 3):306–310
Duman RS (2004) Depression: a case of neuronal life and death? Biol. Psychiatry 56(3):140–145
Ehninger D, Kempermann G (2006) Paradoxical effects of learning the Morris water maze on adult hippocampal neurogenesis in mice may be explained by a combination of stress and physical activity. Genes Brain Beh. 5(1):29–39
Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O (2003) Inflammation is detrimental for neurogenesis in adult brain. Proc. Nat. Acad. Sci. USA. 100(23):13632–13637
el-Defrawy SR, Boegman RJ, Jhamandas K, Beninger RJ (1986) The neurotoxic actions of quinolinic acid in the central nervous system. Can. J. Physiol. Pharmacol. 64(3):369–375
Fitzgerald P, O'Brien SM, Scully P, Rijkers K, Scott LV, Dinan TG (2006) Cutaneous glucocorticoid receptor sensitivity and pro-inflammatory cytokine levels in antidepressant-resistant depression. Psychol. Med. 36(1):37–43
Forlenza MJ, Miller GE (2006) Increased serum levels of 8-hydroxy-2'-deoxyguanosine in clinical depression. Psychosomatic Med. 68(1):1–7
Fuchs E, Czeh B, Kole MH, Michaelis T, Lucassen PJ (2004) Alterations of neuroplasticity in depression: the hippocampus and beyond. Eur. Neuropsychopharmacol. 5(14 Suppl):S481–490
Garthwaite G, Garthwaite J (1987) Quinolinate mimics neurotoxic actions of N-methyl-D-aspartate in rat cerebellar slices. Neurosci. Lett. 79(1–2):35–39
Gould E, McEwen BS, Tanapat P, Galea LA, Fuchs E (1997) Neurogenesis in the dentate gyrus of the adult tree shrew is regulated by psychosocial stress and NMDA receptor activation.J. Neurosci. 17(7):2492–2498
Guillemin GJ, Smith DG, Kerr SJ, Smythe GA, Kapoor V, Armati PJ, Brew BJ (2000) Characterisation of kynurenine pathway metabolism in human astrocytes and implications in neuropathogenesis. Redox Report 5(2–3):108–111
Goshen I, Kreisel T, Ben-Menachem-Zidon O, Licht T, Weidenfeld J, Ben-Hur T, Yirmiya R (2008) Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol Psychiatry 13(7):717–728
Groves JO (2007) Is it time to reassess the BDNF hypothesis of depression? Mol. Psychiatry 12:1079–1088
Halliwell B (2006) Oxidative stress and neurodegeneration: where are we now? J. Neurochem. 97(6):1634–1658 Review
Hand R, Bortone D, Mattar P, Nguyen L, Heng JI, Guerrier S, Boutt E, Peters E, Barnes AP, Parras C, Schuurmans C, Guillemot F, Polleux F (2005) Phosphorylation of neurogenin2 specifies the migration properties and the dendritic morphology of pyramidal neurons in the neocortex. Neuron 48(1):45–62
Henn FA, Vollmayr B (2004) Neurogenesis and depression: etiology or epiphenomenon? Biol. Psychiatry 56(3):146–150
Heyes MP, Saito K, Crowley JS, Davis LE, Demitrack MA, Der M, Dilling LA, Elia J, Kruesi MJ, Lackner A et al (1992) Quinolinic acid and kynurenine pathway metabolism in inflammatory and non-inflammatory neurological disease. Brain 115(Pt 5):1249–1273
Hibbeln JR (1998) Fish consumption and major depression. Lancet 351(9110):1213
Hibbeln JR (2002) Seafood consumption, the DHA content of mothers' milk and prevalence rates of postpartum depression: a cross-national, ecological analysis. J. Affect. Disord. 69(1–3):15–29
Horrobin DF (2001) Phospholipid metabolism and depression: the possible roles of phospholipase A2 and coenzyme A-independent transacylase. Hum. Psychopharmacol. 16(1):45–52
Huang SY, Yang HT, Chiu CC, Pariante CM, Su KP (2006) Omega-3 fatty acids on the forced-swimming test. J. Psychiatr. Res. 42(1):58–63
Ishikawa I, Kitamura H, Kimura K, Saito M (2001) Brain interleukin-1 is involved in blood interleukin-6 response to immobilization stress in rats. Jap. J. Veterinary Res. 49(1):19–25
Kawakita E, Hashimoto M, Shido O (2006) Docosahexaenoic acid promotes neurogenesis in vitro and in vivo. Neurosci. 139(3):991–997
Kempermann G, Neumann H (2003) Microglia: the enemy within? Science 302:1689–1690
Kempermann G, Kuhn HG, Gage FH (1997) More hippocampal neurons in adult mice living in an enriched environment. Nature 386(6624):493–495
Kenis G, Maes M (2002) Effects of antidepressants on the production of cytokines. Int. J. Neuropsychopharmacol. 5(4):401–412 Review
Kent S, Bluthé RM, Kelley KW, Dantzer R (1992) Sickness behavior as a new target for drug development. Trends Pharmacol. Sci. 13(1):24–28
Kerr SJ, Armati PJ, Guillemin GJ, Brew BJ (1998) Chronic exposure of human neurons to quinolinic acid results in neuronal changes consistent with AIDS dementia complex. AIDS 12(4):355–363
Khaspekov L, Kida E, Victorov I, Mossakowski MJ (1989) Neurotoxic effect induced by quinolinic acid in dissociated cell culture of mouse hippocampus. J. Neurosci. Res. 22(2):150–157
Kim YK, Myint AM, Lee BH, Han CS, Lee SW, Leonard BE, Steinbusch HW (2004) T-helper types 1, 2, and 3 cytokine interactions in symptomatic manic patients. Psychiatr. Res. 129(3):267–272
Kim YK, Jung HG, Myint AM, Kim H, Park SH (2007) Imbalance between pro-inflammatory and anti-inflammatory cytokines in bipolar disorder. J. Affect. Disord. 104(1–3):91–95
Kirsch I, Deacon BJ, Huedo-Medina TB, Scoboria A, Moore TJ, Johnson BT (2008) Initial severity and antidepressant benefits: a meta-analysis of data submitted to the Food and Drug Administration. PLoS. Medicine 5(2):e45
Koo JW Duman RS (2008) IL-1beta is an essential mediator of the antineurogenic and anhedonic effects of stress. Proc. Nat. Acad. Sci. USA 105:751–756
Kubera M, Symbirtsev A, Basta-Kaim A, Borycz J, Roman A, Papp M, Claesson M (1996) Effect of chronic treatment with imipramine on interleukin 1 and interleukin 2 production by splenocytes obtained from rats subjected to a chronic mild stress model of depression. Pol. J. Pharmacol. 48:503–506
Kubera M, Van Bockstaele D, Maes M (1999) Leukocyte subsets in treatment-resistant major depression. Pol. J. Pharmacol. 51(6):547–549
Kubera M, Lin AH, Kenis G, Bosmans E, van Bockstaele D, Maes M (2001a) Anti-Inflammatory effects of antidepressants through suppression of the interferon-gamma/interleukin-10 production ratio. J. Clin. Psychopharmacol. 21(2):199–206
Kubera M, Maes M, Holan V, Basta-Kaim A, Roman A, Shani J (2001b) Prolonged desipramine treatment increases the production of interleukin-10, an anti-inflammatory cytokine, in C57BL/6 mice subjected to the chronic mild stress model of depression. J. Affect. Disord. 63(1–3):171–178
Kwidzinski E, Bunse J, Aktas O, Richter D, Mutlu L, Zipp F, Nitsch R, Bechmann I (2005) Indolamine 2,3-dioxygenase is expressed in the CNS and down-regulates autoimmune inflammation. FASEB J. 19(10):1347–1349
Lacosta S, Merali Z, Anisman H (1999) Behavioral and neurochemical consequences of lipopolysaccharide in mice: anxiogenic-like effects. Brain Res. 818(2):291–303
Lapin IP (2003) Neurokynurenines (NEKY) as common neurochemical links of stress and anxiety. Adv. Exp. Med. Biol. 527:121–125
Lestage J, Verrier D, Palin K, Dantzer R (2002) The enzyme indoleamine 2,3-dioxygenase is induced in the mouse brain in response to peripheral administration of lipopolysaccharide and superantigen. Brain Behav. Immun. 16(5):596–601
Levivier M, Przedborski S (1998) Quinolinic acid-induced lesions of the rat striatum: quantitative autoradiographic binding assessment. Neurol. Res. 20(1):46–56
Lin A, Song C, Kenis G, Bosmans E, De Jongh R, Scharpe S, Maes M (2000) The in vitro immunosuppressive effects of moclobemide in healthy volunteers. J. Affect. Disord. 58(1):69–74
Lin PY, Su KP (2007) A meta-analytic review of double-blind, placebo-controlled trials of antidepressant efficacy of omega-3 fatty acids. J. Clin. Psychiatr. 68(7):1056–1061
Machado-Vieira R, Dietrich MO, Leke R, Cereser VH, Zanatto V, Kapczinski F, Souza DO, Portela LV, Gentil V (2007a) Decreased plasma brain derived neurotrophic factor levels in unmedicated bipolar patients during manic episode. Biol. Psychiatry 61(2):142–144
Machado-Vieira R, Andreazza AC, Viale CI, Zanatto V, Cereser V Jr, da Silva Vargas R, Kapczinski F, Portela LV, Souza DO, Salvador M, Gentil V (2007b) Oxidative stress parameters in unmedicated and treated bipolar subjects during initial manic episode: a possible role for lithium antioxidant effects. Neurosci. Lett. 421(1):33–36
Maes M (1993) A review on the acute phase response in major depression. Rev. Neurosci. 4(4):407–416 Review
Maes M (1995) Evidence for an immune response in major depression: a review and hypothesis. Progr. Neuropsychopharmacol. Biol. Psychiatry 19(1):11–38 Review
Maes M (1999) Major depression and activation of the inflammatory response system. Adv. Exp. Med. Biol. 461:25–46 Review
Maes M, Meltzer HY (1995) The Serotonin Hypothesis of major depression. Psychopharmacology, The Fourth Generation of Progress. In: Bloom F, Kupfer D (Ed) Raven Press, pp 933–944
Maes M, Smith RS (1998) Fatty acids, cytokines, and major depression. Biol. Psychiatr. 43(5):313–314
Maes M, De Ruyter M, Claes R, Bosma G, Suy E (1987) The cortisol responses to 5-hydroxytryptophan, orally, in depressive inpatients. J. Affect. Disord. 13(1):23–30
Maes M, De Ruyter M, Claes R, Suy E (1988) Sex-related differences in the relationships between self-rated depression and biological markers. J. Affect. Disord. 15(2):119–125
Maes M, Bosmans E, Suy E, Vandervorst C, De Jonckheere C, Raus J (1990) Immune disturbances during major depression: upregulated expression of interleukin-2 receptors. Neuropsychobiol. 24(3):115–120
Maes M, De Meester I, Vanhoof G, Scharpe S, Bosmans E, Vandervorst C, Verkerk R, Minner B, Suy E, Raus J (1991) Decreased serum dipeptidyl peptidase IV activity in major depression. Biol. Psychiatry 30(6):577–586
Maes M, Bosmans E, Meltzer HY, Scharpe S, Suy E (1993a) Interleukin-1 beta: a putative mediator of HPA axis hyperactivity in major depression? Am. J. Psychiatry 150(8):1189–1193
Maes M, Meltzer HY, Scharpe S, Bosmans E, Suy E, De Meester I, Calabrese J, Cosyns P (1993b) Relationships between lower plasma L-tryptophan levels and immune-inflammatory variables in depression. Psychiatr. Res. 49(2):151–165
Maes M, Meltzer HY, Scharpé S, Cooreman W, Uyttenbroeck W, Suy E, Vandervorst C, Calabrese J, Raus J, Cosyns P (1993c) Psychomotor retardation, anorexia, weight loss, sleep disturbances, and loss of energy: psychopathological correlates of hyperhaptoglobinemia during major depression. Psychiatr. Res. 47(3):229–241
Maes M, Scharpe S, Meltzer HY, Cosyns P (1993d) Relationships between increased haptoglobin plasma levels and activation of cell-mediated immunity in depression. Biol. Psychiatry 34(10):690–701
Maes M, Scharpe S, Meltzer HY, Bosmans E, Suy E, Calabrese J, Cosyns P (1993e) Relationships between interleukin-6 activity, acute phase proteins, and function of the hypothalamic-pituitary-drenal axis in severe depression. Psychiatr. Res. 49(1):11–27
Maes M, Scharpe S, Meltzer H, Okayli G, D'Hondt P, Cosyns P (1994) Increased neopterin and interferon gamma secretion and lower L-tryptophan levels in major depression: further evidence for immune activation in severe depression. Psychiatr. Res. 54:143–160
Maes M, Bosmans E, Calabrese J, Smith R, Meltzer HY (1995a) Interleukin-2 and interleukin-6 in schizophrenia and mania: effects of neuroleptics and mood stabilizers. J. Psychiatry Res. 29(2):141–152
Maes M, Smith R, Scharpe S (1995b) The monocyte-T-lymphocyte hypothesis of major depression. Psychoneuroendocrinol. 20(2):111–116
Maes M, Smith R, Christophe A, Cosyns P, Desnyder R, Meltzer H (1996a) Fatty acid composition in major depression: decreased omega 3 fractions in cholesteryl esters and increased C20: 4 omega 6/C20:5 omega 3 ratio in cholesteryl esters and phospholipids. J. Affect. Disord. 38(1):35–46
Maes M, Wauters A, Verkerk R, Demedts P, Neels H, Van Gastel A, Cosyns P, Scharpe S, Desnyder R (1996b) Lower serum L-tryptophan availability in depression as a marker of a more generalized disorder in protein metabolism. Neuropsychopharmacol. 15(3):243–251
Maes M, Bosmans E, De Jongh R, Kenis G, Vandoolaeghe E, Neels H (1997a) Increased serum IL-6 and IL-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine 9(11):853–858
Maes M, Calabrese J, Jayathilake K, Meltzer HY (1997b) Effects of subchronic treatment with valproate on L-5-HTP-induced cortisol responses in mania: evidence for increased central serotonergic neurotransmission. Psychiatr. Res. 71(2):67–76
Maes M, Delange J, Ranjan R, Meltzer HY, Desnyder R, Cooremans W, Scharpe S (1997c) Acute phase proteins in schizophrenia, mania and major depression: modulation by psychotropic drugs. Psychiatr. Res. 66(1):1–11
Maes M, Vandoolaeghe E, Neels H, Demedts P, Wauters A, Meltzer HY, Altamura C, Desnyder R (1997d) Lower serum zinc in major depression is a sensitive marker of treatment resistance and of the immune/inflammatory response in that illness. Biol. Psychiatry 42(5):349–358
Maes M, Song C, Lin A, De Jongh R, Van Gastel A, Kenis G, Bosmans E, De Meester I, Benoy I, Neels H, Demedts P, Janca A, Scharpe S, Smith RS (1998a) The effects of psychological stress on humans: increased production of pro-inflammatory cytokines and a Th1-like response in stress-induced anxiety. Cytokine 10(4):313–318
Maes M, Song C, Lin A, DeJong R, Van Gastel A, Kenis G, Bosmans E, DeMeester I, Neels H, Janca A, Scharpe S, Smith RS (1998b) Immune and clinical correlates of psychological stress-induced production of interferon-( and IL-10 in humans. In: Plotnikoff NP, Faith RE, Murgo AJ, Good RA (eds) Cytokines, Stress and Immunity, pp 39–50
Maes M, Christophe A, Delanghe J, Altamura C, Neels H, Meltzer HY (1999a) Lowered omega3 polyunsaturated fatty acids in serum phospholipids and cholesteryl esters of depressed patients. Psychiatr. Res. 85(3):275–291
Maes M, Libbrecht I, van Hunsel F, Campens D, Meltzer HY (1999b) Pindolol and mianserin augment the antidepressant activity of fluoxetine in hospitalized major depressed patients, including those with treatment resistance. J. Clin. Psychopharmacol. 19(2):177–182
Maes M, Song C, Lin AH, Bonaccorso S, Kenis G, De Jongh R, Bosmans E, Scharpe S (1999c) Negative immunoregulatory effects of antidepressants: inhibition of interferon-gamma and stimulation of interleukin-10 secretion. Neuropsychopharmacol. 20(4):370–379
Maes M, Song C, Lin AH, Pioli R, Kenis G, Kubera M, Bosmans E (1999d) In vitro immunoregulatory effects of lithium in healthy volunteers. Psychopharmacol. (Berl) 143(4):401–407
Maes M, Van Bockstaele DR, Gastel A, Song C, Schotte C, Neels H, DeMeester I, Scharpe S, Janca A (1999e) The effects of psychological stress on leukocyte subset distribution in humans: evidence of immune activation. Neuropsychobiol. 39(1):1–9
Maes M, Christophe A, Bosmans E, Lin A, Neels H (2000) In humans, serum polyunsaturated fatty acid levels predict the response of proinflammatory cytokines to psychologic stress. Biol. Psychiatr. 47(10):910–920
Maes M, Capuron L, Ravaud A, Gualde N, Bosmans E, Egyed B, Dantzer R, Neveu PJ (2001a) Lowered serum dipeptidyl peptidase IV activity is associated with depressive symptoms and cytokine production in cancer patients receiving interleukin-2-based immunotherapy. Neuropsychopharmacol. 24(2):130–140
Maes M, Ombelet W, De Jongh R, Kenis G, Bosmans E (2001b) The inflammatory response following delivery is amplified in women who previously suffered from major depression, suggesting that major depression is accompanied by a sensitization of the inflammatory response system. J. Affect. Disord. 63(1–3):85–92
Maes M, Ombelet W, Verkerk R, Bosmans E, Scharpe S (2001c) Effects of pregnancy and delivery on the availability of plasma tryptophan to the brain: relationships to delivery-induced immune activation and early post-partum anxiety and depression. Psychol. Med. 31(5):847–858
Maes M, Verkerk R, Bonaccorso S, Ombelet W, Bosmans E, Scharpe S (2002) Depressive and anxiety symptoms in the early puerperium are related to increased degradation of tryptophan into kynurenine, a phenomenon which is related to immune activation. Life Sci. 71(16):1837–1848
Maes M, Mihaylova I, Bosmans E (2007a) Not in the mind of neurasthenic lazybones but in the cell nucleus: patients with chronic fatigue syndrome have increased production of nuclear factor kappa beta. Neuro Endocrinol. Lett. 28(4):456–462
Maes M, Mihaylova I, DeRuyter MD, Kubera M, Bosmans E (2007b) The immune effects of TRYCATs (tryptophan catabolites along the IDO pathway): relevance for depression–and other conditions characterized by tryptophan depletion induced by inflammation. Neuro Endocrinol. Lett. 28(6):826–831
Maes M, Mihaylova I, Kubera M, Bosmans E (2007c) Not in the mind but in the cell: increased production of cyclo-oxygenase-2 and inducible NO synthase in chronic fatigue syndrome. Neuro Endocrinol. Lett. 28(4):463–469
Maes M, Mihaylova I, Leunis JC (2007d) Increased serum IgM antibodies directed against phosphatidyl inositol (Pi) in chronic fatigue syndrome (CFS) and major depression: evidence that an IgM-mediated immune response against Pi is one factor underpinning the comorbidity between both CFS and depression. Neuro Endocrinol. Lett. 28(6):861–867
Maes M, Mihaylova I, Ategis J-C (2008a) Evidence for an IgM-mediated immune response directed against nitro-bovine serum albumin (BSA) in chronic fatigue syndrome (CFS) and major depression (MDD): evidence that the immune response to nitrosative stress-induced damage of BSA is more pronounced in CFS than in MDD. Neuro Endocrinol. Lett. 2008, In press
Maes M, Kubera M, Leunis JC (2008b) The gut-brain barrier in major depression: intestinal mucosal dysfunction with an increased translocation of LPS from gram negative enterobacteria (leaky gut) plays a role in the inflammatory pathophysiology of depression. Neuro Endocrinol. Letters. 29(1):117–124
Malberg JE, Eisch AJ, Nestler EJ, Duman RS (2000) Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 20(24):9104–9110
Mamalakis G, Kalogeropoulos N, Andrikopoulos N, Hatzis C, Kromhout D, Moschandreas J, Kafatos A (2006) Depression and long chain n-3 fatty acids in adipose tissue in adults from Crete. Eur. J. Clin. Nutr. 60(7):882–888
Mancuso M, Coppede F, Migliore L, Siciliano G, Murri L (2006) Mitochondrial dysfunction, oxidative stress and neurodegeneration. J. Alzheimer’s Dis. 10(1):59–73 Review
Marcus SM, Young EA, Kerber KB, Kornstein S, Farabaugh AH, Mitchell J, Wisniewski SR, Balasubramani GK, Trivedi MH, Rush AJ (2005) Gender differences in depression: findings from the STAR*D study. J. Affect. Disord. 87(2–3):141–150
Miller CL, Llenos IC, Dulay JR, Weis S (2006) Upregulation of the initiating step of the kynurenine pathway in postmortem anterior cingulate cortex from individuals with schizophrenia and bipolar disorder. Brain Res. 1073–1074:25–37
Mohr DC, Goodkin DE, Islar J, Hauser SL, Genain CP (2001) Treatment of depression is associated with suppression of nonspecific and antigen-specific T(H)1 responses in multiple sclerosis. Arch. Neurol. 58(7):1081–1086
Moncada S, Bolanos JP (2006) Nitric oxide, cell bioenergetics and neurodegeneration. J. Neurochem. 97(6):1676–1689 Review
Monje ML, Toda H, Palmer TD (2003) Inflammatory blockade restores adult hippocampal neurogenesis. Science 302:1760–1765
Monteggia LM, Luikart B, Barrot M, Theobold D, Malkovska I, Nef S, Parada LF, Nestler EJ (2007) Brain-derived neurotrophic factor conditional knockouts show gender differences in depression-related behaviors. Biol. Psychiatr. 61(2):187–197
Muller N, Schwarz MJ, Dehning S, Douhe A, Cerovecki A, Goldstein-Muller B, Spellmann I, Hetzel G, Maino K, Kleindienst N, Moller HJ, Arolt V, Riedel M (2006) The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol. Psychiatr. 11(7):680–684
Musselman DL, Lawson DH, Gumnick JF, Manatunga AK, Penna S, Goodkin RS, Greiner K, Nemeroff CB, Miller AH (2001) Paroxetine for the prevention of depression induced by high-dose interferon alfa. New Engl. J. Med. 344(13):961–966
Myint AM, Kim YK, Verkerk R, Park SH, Scharpe S, Steinbusch HW, Leonard BE (2007) Tryptophan breakdown pathway in bipolar mania. J. Affect. Disord. 102(1–3):65–72
Ngai LY, Herbert J (2005) Glucocorticoid enhances the neurotoxic actions of quinolinic acid in the striatum in a cell-specific manner. J. Neuroendocrinol. 17(7):424–434
Nguyen KT, Deak T, Owens SM, Kohno T, Fleshner M, Watkins LR, Maier SF (1998) Exposure to acute stress induces brain interleukin-1beta protein in the rat. J. Neurosci. 18:2239–2246
Noraberg J, Poulsen FR, Blaabjerg M, Kristensen BW, Bonde C, Montero M, Meyer M, Gramsbergen JB, Zimmer J (2005) Organotypic hippocampal slice cultures for studies of brain damage, neuroprotection and neurorepair. Curr. Drug Targets. CNS Neurol. Disord. 4(4):435–452 Review
Noraberg J, Jensen CV, Bonde C, Montero M, Nielsen JV, Jensen NA, Zimmer J (2007) Developmental expression of fluorescent proteins in organotypic hippocampal slice cultures from transgenic mice with example of excitotoxic neurodegeneration. ATLA. 35(1):61–70 Review
O'Brien SM, Scully P, Fitzgerald P, Scott LV, Dinan TG (2007) Plasma cytokine profiles in depressed patients who fail to respond to selective serotonin reuptake inhibitor therapy. J. Psychiatr. Res. 41(3–4):326–331
O'Connor JC, Lawson MA, André C, Moreau M, Lestage J, Castanon N, Kelley KW, Dantzer R (2008) Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol. Psychiatry. [Epub ahead of print]
Overstreet DH, Friedman E, Mathé AA, Yadid G (2005) The Flinders Sensitive Line rat: a selectively bred putative animal model of depression. Neurosci. Biobeh. Rev. 29(4–5):739–759
Pariante CM, Miller AH (2001) Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol. Psychiatr. 49(5):391–404
Patel HC, Ross FM, Heenan LE, Davies RE, Rothwell NJ, Allan SM (2006) Neurodegenerative actions of interleukin-1 in the rat brain are mediated through increases in seizure activity. J. Neurosci. Res. 83(3):385–391
Peet M, Murphy B, Shay J, Horrobin D (1998) Depletion of omega-3 fatty acid levels in red blood cell membranes of depressive patients. Biol. Psychiatr. 43(5):315–319
Pemberton LA, Kerr SJ, Smythe G, Brew BJ (1997) Quinolinic acid production by macrophages stimulated with IFNγ, TNF-alpha, and IFNα. J. Interf. Cytokine Res. 17(10):589–595
Article CAS Google Scholar
Periyasamy S, Sanchez ER (2002) Antagonism of glucocorticoid receptor transactivity and cell growth inhibition by transforming growth factor-beta through AP-1-mediated transcriptional repression. Int. J. Biochem. Cell Biol. 34(12):1571–1585
Pettit JW, Lewinsohn PM, Joiner TE Jr (2006) Propagation of major depressive disorder: relationship between first episode symptoms and recurrence. Psychiatr. Res. 141(3):271–278
Pláteník J, Stopka P, Vejrazka M, Stípek S (2001) Quinolinic acid-iron(ii) complexes: slow autoxidation, but enhanced hydroxyl radical production in the Fenton reaction. Free Radical Res. 34(5):445–459
Polleux F, Ghosh A (2002) The slice overlay assay: a versatile tool to study the influence of extracellular signals on neuronal development. Science STKE 136(19):1–11
Pong K (2003) Oxidative stress in neurodegenerative diseases: therapeutic implications for superoxide dismutase mimetics. Exp. Opin. Biol. Therap. 3(1):127–139 Review
Post RM (1992) Transduction of psychosocial stress into the neurobiology of recurrent affective disorder. Am. J. Psychiatry 149(8):999–1010
Post RM (2007) Kindling and sensitization as models for affective episode recurrence, cyclicity, and tolerance phenomena. Neurosci. Biobehav. Rev 31(6):858–873
Potashkin JA, Meredith GE (2006) The role of oxidative stress in the dysregulation of gene expression and protein metabolism in neurodegenerative disease. Antioxidant Redox Sign. 8(1–2):144–151 Review
Qian L, Hong JS, Flood PM (2006) Role of microglia in inflammation-mediated degeneration of dopaminergic neurons: neuroprotective effect of interleukin 10. J. Neural Transm. Suppl. 70:367–371
Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, Knapp DJ, Crews FT (2007) Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55(5):453–462
Rao JS, Lee HJ, Rapoport SI, Bazinet RP (2008) Mode of action of mood stabilizers: is the arachidonic acid cascade a common target? Mol. Psychiatr. 13(6):585–596
Rios C, Santamaria A (1991) Quinolinic acid is a potent lipid peroxidant in rat brain homogenates. Neurochem. Res. 16(10):1139–1143
Sakic B, Szechtman H, Braciak T, Richards C, Gauldie J, Denburg JA (1997) Reduced preference for sucrose in autoimmune mice: a possible role of interleukin-6. Brain Res. Bull. 44(2):155–165
Sakic B, Gauldie J, Denburg JA, Szechtman H (2001) Behavioral effects of infection with IL-6 adenovector. Brain Beh Immun. 15(1):25–42
Sandi C (2004) Stress, cognitive impairment and cell adhesion molecules. Nature Reviews Neuroscience 5:917–930
Sandi C, Bisaz R (2007) A model for the involvement of neural cell adhesion molecules in stress-related mood disorders. Neuroendocrinol. 85(3):158–176
Sapolsky RM (2004) Is impaired neurogenesis relevant to the affective symptoms of depression? Biol. Psychiatry 56(3):137–139
Sarandol A, Sarandol E, Eker SS, Erdinc S, Vatansever E, Kirli S (2007) Major depressive disorder is accompanied with oxidative stress: short-term antidepressant treatment does not alter oxidative-antioxidative systems. Hum. Psychopharmacol. 22(2):67–73
Schiepers OJ, Wichers MC, Maes M (2005) Cytokines and major depression. Prog Neuropsychopharmacol. Biol. Psychiatr. 29(2):201–217
Schmidt HD, Duman RS (2007) The role of neurotrophic factors in adult hippocampal neurogenesis, antidepressant treatments and animal models of depressive-like behavior. Beh. Pharmacol. 18(5–6):391–418
Schulte-Herbruggen O, Nassenstein C, Lommatzsch M, Quarcoo D, Renz H, Braun A (2005) Tumor necrosis factor-alpha and interleukin-6 regulate secretion of brain-derived neurotrophic factor in human monocytes. J. Neuroimmunol. 160(1–2):204–209
Schwarcz R, Köhler C (1983) Differential vulnerability of central neurons of the rat to quinolinic acid. Neurosci. Lett. 38(1):85–90
Shapira-Lichter I, Beilin B, Ofek K, Bessler H, Gruberger M, Shavit Y, Seror D, Grinevich G, Posner E, Reichenberg A, Soreq H, Yirmiya R (2008) Cytokines and cholinergic signals co-modulate surgical stress-induced changes in mood and memory. Brain Beh. Immun. 22:388–398
Smith MA, Makino S, Kvetnansky R, Post RM (1995) Stress and glucocorticoids affect the expression of brain-derived neurotrophic factor and neurotrophin-3 mRNAs in the hippocampus. J. Neurosci. 15:1768–1777
Sobocki P, Jonsson B, Angst J, Rehnberg C (2006) Cost of depression in Europe. J. Ment. Health Policy Econ. 9(2):87–98
Sobczak S, Honig A, Christophe A, Maes M, Helsdingen RW, De Vriese SA, Riedel WJ (2004) Lower high-density lipoprotein cholesterol and increased omega-6 polyunsaturated fatty acids in first-degree relatives of bipolar patients. Psychol. Med 34(1):103–112
Song C, Leonard BE (1995) Interleukin-2-induced changes in behavioural, neurotransmitter, and immunological parameters in the olfactory bulbectomized rat. Neuroimmunomodulation. 2(5):263–273
Song C, Leonard BE, Horrobin DF (2004) Dietary ethyl-eicosapentaenoic acid but not soybean oil reverses central interleukin-1-induced changes in behavior, corticosterone and immune response in rats. Stress 7(1):43–54
Article PubMed CAS Google Scholar
Song C, Li X, Kang Z, Kadotomi Y (2007) Omega-3 fatty acid ethyl-eicosapentaenoate attenuates IL-1beta-induced changes in dopamine and metabolites in the shell of the nucleus accumbens: involved with PLA2 activity and corticosterone secretion. Neuropsychopharmacol. 32(3):736–744
Steptoe A, Hamer M, Chida Y (2007) The effects of acute psychological stress on circulating inflammatory factors in humans: a review and meta-analysis. Brain Beh. Immun. 21:901–912
Stockmeier CA, Mahajan GJ, Konick LC, Overholser JC, Jurjus GJ, Meltzer HY, Uylings HB, Friedman L, Rajkowska G (2004) Cellular changes in the postmortem hippocampus in major depression. Biol. Psychiatr. 56(9):640–650
Stone TW, Behan WM (2007) Interleukin-1beta but not tumor necrosis factor-alpha potentiates neuronal damage by quinolinic acid: protection by an adenosine A2A receptor antagonist. J. Neurosci. Res. 85(5):1077–1085
Stork O, Welzl H, Wotjak CT, Hoyer D, Delling M, Cremer H, Schachner M (1999) Anxiety and increased 5-HT1A receptor response in NCAM null mutant mice. J. Neurobiol. 40:343–355
Turner CA, Akil H, Watson SJ, Evans SJ (2006) The fibroblast growth factor system and mood disorders. Biol. Psychiatr. 59(12):1128–1135
Ueda S, Sakakibara S, Yoshimoto K (2005) Effect of long-lasting serotonin depletion on environmental enrichment-induced neurogenesis in adult rat hippocampus and spatial learning. Neurosci. 135:395–402
Vaidya VA, Duman RS (2001) Depression-emerging insights from neurobiology. Brit. Med. Bull. 57:61–79
van Praag H, Kempermann G, Gage FH (1999) Running increases cell proliferation and neurogenesis in the adult mouse dentate gyrus. Nature Neurosci. 2(3):266–270
Viviani B, Gardoni F, Bartesaghi S, Corsini E, Facchi A, Galli CL, Di Luca M, Marinovich M (2006) Interleukin-1 beta released by gp120 drives neural death through tyrosine phosphorylation and trafficking of NMDA receptors. J. Biol. Chem. 281(40):30212–3022
Wadee AA, Kuschke RH, Wood LA, Berk M, Ichim L, Maes M (2002) Serological observations in patients suffering from acute manic episodes. Human Psychopharmacol. 17(4):175–179
Walz JC, Frey BN, Andreazza AC, Cereser KM, Cacilhas AA, Valvassori SS, Quevedo J, Kapczinski F (2007) Effects of lithium and valproate on serum and hippocampal neurotrophin-3 levels in an animal model of mania. J. Psychiatr. Res. 42(5):416–421
Wang JY, Wen LL, Huang YN, Chen YT, Ku MC (2006) Dual effects of antioxidants in neurodegeneration: direct neuroprotection against oxidative stress and indirect protection via suppression of glia-mediated inflammation. Curr. Pharmac. Design 12(27):3521–3533 Review
Wichers MC, Maes M (2004) The role of indoleamine 2, 3-dioxygenase (IDO) in the pathophysiology of interferon-alpha-induced depression. J. Psychiatr. Neurosci. 29(1):11–17
Wichers MC, Koek GH, Robaeys G, Verkerk R, Scharpe S, Maes M (2005) IDO and interferon-alpha-induced depressive symptoms: a shift in hypothesis from tryptophan depletion to neurotoxicity. Mol. Psychiatr. 10(6):538–544
Wichers MC, Kenis G, Koek GH, Robaeys G, Nicolson NA, Maes M (2007) Interferon-alpha-induced depressive symptoms are related to changes in the cytokine network but not to cortisol. J. Psychosom. Res. 62(2):207–214
Wu A, Ying Z, Gomez-Pinilla F (2004) Dietary omega-3 fatty acids normalize BDNF levels, reduce oxidative damage, and counteract learning disability after traumatic brain injury in rats. J. Neurotrauma. 21:1457–1467
Xia Z, DePierre JW, Nassberger L (1996) Tricyclic antidepressants inhibit IL-6, IL-1 beta and TNF-alpha release in human blood monocytes and IL-2 and interferon-gamma in T cells. Immunopharmacol. 34(1):27–37
Xu Y, Ku B, Cui L, Li X, Barish PA, Foster TC, Ogle WO (2007) Curcumin reverses impaired hippocampal neurogenesis and increases serotonin receptor 1A mRNA and brain-derived neurotrophic factor expression in chronically stressed rats. Brain Res. 1162:9–18
Yang R, Han X, Uchiyama T, Watkins SK, Yaguchi A, Delude RL, Fink MP (2003) IL-6 is essential for development of gut barrier dysfunction after hemorrhagic shock and resuscitation in mice. Am. J. Physiol. Gastrointestinal Liver Physiol. 285(3):G621–629
Yirmiya R (1996) Endotoxin produces a depressive-like episode in rats. Brain Res. 711(1–2):163–74
Yirmiya R (1997) Behavioral and psychological effects of immune activation: implications for ‘depression due to a general medical condition’. Curr. Opin. Psychiatr. 10:470–476
Yirmiya R, Weidenfeld J, Pollak Y, Morag M, Morag A, Avitsur R, Barak O, Reichenberg A, Cohen E, Shavit Y, Ovadia H (1999) Cytokines, "depression due to a general medical condition," and antidepressant drugs. Adv. Exp. Med. Biol. 461:283–316
Yirmiya R, Pollak Y, Barak O, Avitsur R, Ovadia H, Bette M, Weihe E, Weidenfeld J (2001) Effects of antidepressant drugs on the behavioral and physiological responses to lipopolysaccharide (LPS) in rodents. Neuropsychopharmacol. 24(5):531–544
Zhu SW, Pham TM, Aberg E, Brene S, Winblad B, Mohammed AH, Baumans V (2006) Neurotrophin levels and behaviour in BALB/c mice: impact of intermittent exposure to individual housing and wheel running. Beh. Brain Res. 167(1):1–8
Zou JY, Crews FT (2005) TNF alpha potentiates glutamate neurotoxicity by inhibiting glutamate uptake in organotypic brain slice cultures: neuroprotection by NF kappa B inhibition. Brain Res. 1034(1–2):11–24
Download references
Author information
Authors and affiliations.
Clinical Research Center for Mental Health, Olmenlaan 9, Antwerp Wilrijk, 2610, Belgium
Michael Maes
Department of Psychology, The Hebrew University, Jerusalem, Israel
Raz Yirmyia
Molecular Neurobiology Laboratory, Medical Biotechnology Center, University of Southern Denmark, Odense, Denmark
Jens Noraberg
Department of Anatomy and Neurobiology, University of Southern Denmark, Odense, Denmark
Department of Neurobiology, Care Sciences and Society, Karolinska University Hospital, MR Center, Stockholm, Sweden
Stefan Brene
Section on Nutritional Neurochemistry, LMBB, DICBR, NIAAA, NIH, Washington, DC, USA
Joe Hibbeln
Department of Psychiatry, University of Padova, Padova, Italy
Giulia Perini
Institute of Pharmacology, Polish Academy of Science, Krakow, Poland
Marta Kubera
Psychiatric Department, 1st Medical Faculty Charles University in Prague, Prague, Czech Republic
Hadassah Biological Psychiatry Laboratory, Hadassah-Hebrew University, Jerusalem, Israel
Bernard Lerer
Department of Psychiatry, University of Naples SUN, Naples, Italy
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to Michael Maes .
Rights and permissions
Reprints and permissions
About this article
Maes, M., Yirmyia, R., Noraberg, J. et al. The inflammatory & neurodegenerative (I&ND) hypothesis of depression: leads for future research and new drug developments in depression. Metab Brain Dis 24 , 27–53 (2009). https://doi.org/10.1007/s11011-008-9118-1
Download citation
Received : 02 April 2008
Accepted : 28 October 2008
Published : 16 December 2008
Issue Date : March 2009
DOI : https://doi.org/10.1007/s11011-008-9118-1
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Inflammation
- Neurodegeneration
- Oxidative stress
- Nitrosative stress
- Find a journal
- Publish with us
- Track your research

A Story of Depression as a Disease
How we think about depression impacts clinical treatment..
Updated January 3, 2024 | Reviewed by Monica Vilhauer
- What Is Depression?
- Find a therapist to overcome depression
- A model of depression as a disease is incomplete and misleading. It leads to suboptimal treatment choices.
- The "separation distress hypothesis" is an alternative model. It is an integrated, causal model of depression.
- This alternative model leads to psychologically meaningful treatments with higher efficacy than SSRIs.
Is there a difference between these statements: “I have depression ” and "I am depressed?" The first sentence describes something lasting, possibly chronic. The second one describes the state of the person's brain and mind.
As Jonathan Shedler pointed out, the first description provides an illusion of an explanation of why the person is distressed : “Why do I feel this way? Because I have an illness called “depression.”
Can we pause here for a moment? This is not an accident that we talk about depression as a disease. We have been taught to do so. How?
Programming by language happens all the time in our culture. For example, we speak Starbucks: “I’d like a grande-soy-latte, please.”
In Chapter 4 of Ethan Watters’ book [1], McGill University professor Laurence Kirmayer and Keio University Professor Junko Kitanaka shared a remarkable account of how one of the stories of depression was written in Japan [1]. Before the 1990s, there was no word in Japanese for mild-to-moderate depression; and the sales of selective serotonin reuptake inhibitors (SSRIs) were zero, while the sales were approximately 13 billion dollars per year in other countries combined. Then, GlaxoSmithKline, the manufacturer of Paxil, started a massive marketing campaign. Two of their campaign's key messages were that depression was a common disease caused by a chemical imbalance in the brain and that “antidepressants” restored the balance. As a result, the sales of Paxil in Japan reached the level of a billion dollars per year by 2008.
One of the components of this story was the term “antidepressant,” which has been used in the USA since the 1950s. This term is attributed to Max Lurie, a psychiatrist in Cincinnati [2]. He referred to isoniazid , a monoamine oxidase inhibitor (MAOI) as an “antidepressant.” Thereafter, all pills prescribed for depression have been called “antidepressants,” including selective serotonin reuptake inhibitors (SSRIs). In labeling isoniazid as an “antidepressant,” Dr. Lurie did something quite customary in medicine (consider anti-inflammatory or antinausea medications). However, he was influenced by the story that depression was a disease, which led him to use medical terminology.
The prefix “anti” creates an impression of a dichotomous system, where the disease drives the pathological process forward, while an “anti-depressant” drives it back to health.
Here is what I think happens when people hear the term antidepressant : “There is a clear-cut thing, called depression. Depression is a disease, caused by a chemical imbalance in the brain. A cure from this disease is an “antidepressant” – it restores the chemical balance.”
Then, there is a twist:
“When your antidepressant didn’t work, we will consider your depression to be ' treatment-resistant .' Why? Because the anti-depressant was supposed to work. The very name suggests so. The only reason why it didn’t work was that your unruly depression was 'resistant.' Then what? Electro-convulsive therapy (ECT). Still resistant? Ketamine . Resistant still? Surgery.”
Please note that “treatment-resistant depression” was defined in the 1970s as a person’s lack of symptomatic improvement in response to two different courses of “antidepressants” [3]. Therefore, the term “treatment-resistant depression” assumes that anti-depressants are supposed to work. Such a definition is an example of circular logic – you define a drug based on the disease, and then you redefine the disease based on the patient's reaction to the drug.
Now we know that there is no causal theory of serotonin imbalance leading to depressive symptoms [9], and the evidence of the efficacy of SSRIs for depression is weak – it is three times lower than that of psychodynamic psychotherapy [4].
If the tale of depression as a disease is misleading, what are the alternatives? Jonathan Shedler suggested a useful metaphor – a fever. Fever is a non-specific state – it is common in various conditions. Flu, on the other hand, is a disease and we know its etiology (a set of causes) – it is a viral infection. Shedler suggested that depression, like fever, is a non-specific state, not a disease.
We know that Tylenol treats the symptom (fever) but does not cure the flu. Treating symptoms is important [5], as we could die from hyperthermia, but we need to know that the immune system cures the flu, not the Tylenol. Therefore, it would be misleading to describe a lasting flu as “treatment resistant” based on it not responding to Tylenol.
In addition to Shedler’s metaphor of a fever, an integrated, causal model of depression as a state was proposed in 2009 [10], and then further elaborated and refined [6, 9]. This model comes from Affective Neuroscience by the late Jaak Panksepp and his colleague Douglas Watt, as well as Mark Solms, Maggie Zellner, and others [6, 9]. It is called the "separation distress hypothesis." Some of the ideas in this model go back to Sigmund Freud and John Bowlby , but Pansepp and Watt made critical multidisciplinary contributions.

Summarizing the separation distress hypothesis here would not do it justice, as it is reasonably complex (so is the phenomenon it represents). You can find the latest update and systematic review of this model here [9]. The separation distress hypothesis combines innate, developmental, biological, psychological, and environmental factors. It does not reduce the macro phenomenon of depression to a molecular level of serotonin while ignoring all the levels in between. By now we have accumulated considerable evidence from multiple perspectives in support of the separation distress hypothesis [see 9 for a comprehensive review, as well as 6 and 8].
Repeated experiences of neglect, abuse, or abandonment in childhood , maladaptive habits, sleep disturbance, acute traumas at any age, complex trauma, and other factors can all lead to the patient suffering from repeated episodes (states) of depression. As you can see, depression, like fever, is non-specific and there are many possible pathways to it. It is biological and psychological at the same time.
What might the shift to the separation distress hypothesis result in? First, the dominant chemical imbalance story would have to be de-prioritized. An alternative understanding described by Mark Solms and his colleagues is that the feeling of depression means something [6,7]. Solms reminds us that this is not a new idea in medicine. A sensation of acute pain in the leg means that there is possibly a laceration there. Nausea means a possibly upset stomach. These “messages” guide us to what the problem is and where.
One of the meanings of depression, according to Mark Solms, is that our normal need to feel cared for is unmet [6, 7]. We feel painfully alone, unattached, or abandoned. This message is something we can notice, acknowledge, and work with in psychotherapy. Further, there are meaningful psychological reasons why the patient feels depressed, episodically or chronically. There is a way to discover in psychotherapy how this state came about and then work together with the patient to get to a stable resolution of this problem.
Using an etiological approach to depression would allow us to focus on the causal treatment that has shown significantly higher efficacy than SSRIs [4].
It is worthwhile saying that the separation distress hypothesis, like any other theory, has some limitations. For example, it lacks cultural sensitivity. However, I believe that this etiology-based, integrated model is more beneficial in guiding treatment choices for depression than the "chemical imbalance" model.
[1] Watters, E. (2010). Crazy like us: The globalization of the American psyche. Simon and Schuster.
[2] Pereira, V. S., & Hiroaki-Sato, V. A. (2018). A brief history of antidepressant drug development: from tricyclics to beyond ketamine. Acta neuropsychiatrica, 30(6), 307-322.
[3] Murphy JA, Sarris J, Byrne GJ. A review of the conceptualisation and risk factors associated with treatment-resistant depression. Depress Res Treat. 2017;2017:4176825. doi:10.1155/2017/4176825
[4] Shedler, J. (2010). The efficacy of psychodynamic psychotherapy. American psychologist, 65(2), 98.
[5] Solms, M. (2018). The scientific standing of psychoanalysis. BJPsych International, 15(1), 5-8.
[6] Zellner, M. R., Watt, D. F., Solms, M., & Panksepp, J. (2011). Affective neuroscientific and neuropsychoanalytic approaches to two intractable psychiatric problems: why depression feels so bad and what addicts really want. Neuroscience & Biobehavioral Reviews, 35(9), 2000-2008.
[7] Solms, M. L. (2018). The neurobiological underpinnings of psychoanalytic theory and therapy. Frontiers in Behavioral Neuroscience, 12, 402180.
[8] Blomstedt, P., Hariz, M. I., Lees, A., Silberstein, P., Limousin, P., Yelnik, J., & Agid, Y. (2008). Acute severe depression induced by intraoperative stimulation of the substantia nigra: a case report. Parkinsonism & related disorders, 14(3), 253-256.
[9] Watt, D. F. (2023). The separation distress hypothesis of depression–an update and systematic review. Neuropsychoanalysis, 1-57.
[10] Watt, D. F., & Panksepp, J. (2009). Depression: An evolutionarily conserved mechanism to terminate protracted separation distress. A review of aminergic, peptidergic and neural network perspectives. (Target article with invited commentaries). Neuropsychoanalysis, 11(1), 7–51. https://doi.org/10.1080/15294145.2009.10773593

Alexey Tolchinsky, Psy.D. , is a clinical psychologist in private practice in Maryland and a Clinical Fellow of the Neuropsychoanalysis Association.
- Find a Therapist
- Find a Treatment Center
- Find a Psychiatrist
- Find a Support Group
- Find Teletherapy
- United States
- Brooklyn, NY
- Chicago, IL
- Houston, TX
- Los Angeles, CA
- New York, NY
- Portland, OR
- San Diego, CA
- San Francisco, CA
- Seattle, WA
- Washington, DC
- Asperger's
- Bipolar Disorder
- Chronic Pain
- Eating Disorders
- Passive Aggression
- Personality
- Goal Setting
- Positive Psychology
- Stopping Smoking
- Low Sexual Desire
- Relationships
- Child Development
- Therapy Center NEW
- Diagnosis Dictionary
- Types of Therapy

Understanding what emotional intelligence looks like and the steps needed to improve it could light a path to a more emotionally adept world.
- Coronavirus Disease 2019
- Affective Forecasting
- Neuroscience
- Open access
- Published: 16 February 2021
On inflammatory hypothesis of depression: what is the role of IL-6 in the middle of the chaos?
- Elnaz Roohi 1 ,
- Nematollah Jaafari 2 &
- Farshad Hashemian 1
Journal of Neuroinflammation volume 18 , Article number: 45 ( 2021 ) Cite this article
13k Accesses
80 Citations
16 Altmetric
Metrics details
Many patients with major depressive disorder (MDD) are reported to have higher levels of multiple inflammatory cytokines including interleukin 6 (IL-6). Recent studies both pre-clinical and clinical have advocated for the functional role of IL-6 in development of MDD and suggested a great potential for targeting this cytokine to open new avenues in pharmacotherapy of depression. The purpose of the present narrative review was to provide an integrated account of how IL-6 may contribute to development of depression. All peer-reviewed journal articles published before July 2020 for each area discussed were searched by WOS, PubMed, MEDLINE, Scopus, Google Scholar, for original research, review articles, and book chapters. Publications between 1980 and July 2020 were included. Alterations in IL-6 levels, both within the periphery and the brain, most probably contribute to depression symptomatology in numerous ways. As IL-6 acts on multiple differing target tissues throughout the body, dysregulation of this particular cytokine can precipitate a multitude of events relevant to depression and blocking its effects can prevent further escalation of inflammatory responses, and potentially pave the way for opening new avenues in diagnosis, treatment, and prevention of this debilitating disorder.
Major depressive disorder (MDD) is a leading cause of disability throughout the world with a global prevalence of 2.6–5.9% [ 1 ]. The total estimated number of people living with depression worldwide increased by 49.86% from 1990 to 2017 [ 2 ]. According to worldwide projections, MDD will be the single major cause of burden of all health conditions by 2030 [ 3 ]. MDD is characterized by periods of low mood, altered cognition, considerable functional burden including impaired occupational functioning and psychosocial disability [ 4 ]. Despite available pharmacotherapeutic options, 30–60% of patients with MDD are not responsive to available treatments [ 5 ] and the rate of remission of the disease is often < 50% [ 6 ], while recurrence rates are more than 85% within 10 years of a depressive episode, and average about ≥ 50% within 6 months of assumed clinical remission [ 4 ]. Indeed, there exists no compelling evidence that current treatments are capable of disease modification in MDD patients. Thus, therapeutic deficiency in treatment outcomes reflects the demand for revitalizing psychiatric therapeutics with novel pharmacotherapeutic options that engage non-monoaminergic molecular targets.
A large body of evidence suggests that inflammation has central role in pathogenesis of MDD [ 7 , 8 , 9 , 10 , 11 , 12 , 13 ]. However, the exact mechanisms underlying inflammation-induced depression are not completely elucidated [ 3 ]. Historically, the “monoamine-depletion hypothesis” has been the main proposed pathophysiology [ 14 ]; nevertheless, this hypothesis alone cannot fully account for pathogenesis of MDD [ 15 , 16 ]. In recent years, “inflammatory hypothesis” has been proposed [ 17 ]. However, it is noteworthy that it was probably in the early 1990s that for the first time, possible relationships between the peripheral immune system and major depression was studied [ 18 ]. Maes et al. (1992) established immune cell profile of patients with depression and advocated for the existence of a systemic immune activation during major depressive disorder [ 19 ]. Moreover, correlations between IL-6 activity, acute phase proteins, and hyperactivity of the hypothalamic-pituitary-adrenal (HPA) axis were suggested in severe depression [ 20 ].
Most proximally, inflammation is regulated by expression of immune response genes including interleukin (IL)-1B, tumor necrosis factor (TNF), and IL-6 which promote secretion of pro-inflammatory cytokines leading to systemic inflammation. Distally, inflammation is regulated in the brain where socio-environmental cues including possible threat are detected. This neuro-inflammatory link can activate the conserved transcriptional response to adversity (CTRA) before happening of a possible threat or bacterial infection. However, the negative aspect of central regulation of systemic inflammation is that it can give social and foreseen dangers (including those that have not yet occurred or may never actually happen) the ability to activate the CTRA in the absence of actual physical danger. Under normal conditions, CTRA-related inflammatory activity is downregulated by the HPA axis via the production of cortisol. Nevertheless, when prolonged actual or perceived social threat or physical danger is present, glucocorticoid resistance can develop which leads to excessive inflammation that heightens a person’s risk for development of several disorders including MDD, especially if activation of these pathways is prolonged [ 21 ]. As mentioned above, the current understanding of MDD encloses not only alterations in neurotransmitters, but also changes in immune and endocrine functioning as well as neural circuits [ 22 ]. This broadened framework has just started to inform a wide array of novel, personalized therapeutics that are showcasing great promise in a new holistic approach to MDD [ 23 ].
Cytokines are implicated in pathogenesis of MDD [ 24 , 25 , 26 , 27 , 28 , 29 , 30 ]. Risk factors of developing MDD include familial, developmental, psychological, and medical risk factors as well as molecular factors associated with genetics, epigenetics, gene expression, and also those related to the endocrine and the immune system [ 31 , 32 ]. All these risk factors have been shown to be related with changes in cytokine production or signaling. In other words, cytokines are involved in almost every predisposing or precipitating risk factor associated with MDD [ 24 ]. Indeed, there is accumulating evidence in favor of involvement of pro-inflammatory cytokines in pathophysiology of depression [ 24 , 29 , 33 , 34 , 35 , 36 ]. Various studies reported higher levels of multiple inflammatory markers including IL-6 in patients with MDD [ 37 , 38 , 39 , 40 , 41 ]. Of all pro-inflammatory cytokines, changes in IL-6 serum levels have been reported as one of the most reproducible abnormalities in MDD [ 38 ].
The aim of the present narrative review is to elucidate the fundamentals, implications, challenges of cytokine research specifically IL-6 in major depressive disorder. This comprises of the following:
-) A Brief overview of cytokines
-) Cytokine categories according to immunological function.
-) IL-6 as a pleiotropic cytokine.
-) Brief overview of chemokines and their role in Depression.
-) Challenges of cytokine research in psychiatry.
-) IL-6 alterations in depression.
-) Effects of IL-6 on neurotransmitters’ synthesis, signaling, metabolism, and function.
-) Effects of IL-6 levels on brain morphology in depression.
-) Blockade of IL-6 and its receptor in the periphery as a potential therapeutic option in MDD.
-) Possible role of IL-6 together with gut microbiota in pathogenesis of depression.
-) Elevated levels of IL-6 in patients with COVID-19 infection.
The present article is a narrative review. All peer-reviewed journal articles published before July 2020 for each area discussed were searched by WOS, PubMed, MEDLINE, Scopus, Google Scholar, for original research, review articles, and book chapters. We selected articles on the basis of being comprehensive, innovative, and informative for an in-depth understanding and a critical debate on the topic. Publications between 1973 and 2020 were included.
A brief overview of cytokines
Cytokines are a broad category of released proteins that act as signaling molecules to regulate inflammation and cellular activities [ 24 , 42 ]. They are produced by different immune cells (e.g., macrophages, lymphocytes, mast cells), parenchymal cells, endothelial and epithelial cells, fibroblasts, adipocytes, and stromal cells within the periphery [ 24 , 43 ]. Additionally, they are produced by microglia, astrocytes, and neurons in the brain [ 44 ]. Cytokines from the periphery (peripherally produced cytokines) can exert influences on inflammatory processes in the brain [ 45 , 46 ]. Indeed, they can enter blood-brain barrier (BBB) and affect the brain via humoral (accessing the brain through leaky secretions of the BBB such as choroid plexus), neural (through stimulation of primary afferent nerve fibers in the vagus nerve), and cellular (through stimulation of microglia by pre-inflammatory cytokines to produce monocyte chemottractant protein-1 and recruit monocytes to the brain) pathways [ 47 ]. Most cytokines function in their immediate microenvironment. Few of them are involved in paracrine signaling which indeed is fundamental to the control of an inflammatory response within a given tissue or organ and the activation of a coordinated immune response that involves multiple cell types [ 48 ]. Apart from navigating the immune system to defend the body from pathogens, cytokines have a modifying effect on neurotransmission [ 49 ].
It’s also noteworthy that the same cytokines can be produced by multiple cell types. For example, white blood cells, endothelium, fat cells, and other cells can produce TNF-α [ 50 ]. Additionally, one single cell can release different cytokines. For instance, T Helper type 2 (T H 2) cells can produce IL-3, IL-4, IL-5, IL-6, and IL-13 [ 24 ]. Cytokines can have pleiotropic, redundant, synergistic, and antagonistic effects [ 51 ]. The phenomenon that a single cytokine can act on several different cell types is called pleiotropy [ 51 ]. For instance, IL-10 can activate T H 2 cells and B cells, yet inhibit macrophages and T helper type 1 (T H 1) cells. Thus, being immunostimulatory as well as being immunosuppressive [ 52 ]. Cytokines are redundant in their activity, i.e., similar functions can be exerted by different cytokines. For instance, interferon (IFN)-γ, IL-2, and TNF-α enhance cellular immunity and production of cytotoxic cell contacts [ 53 ]. Cytokines can also act synergistically, i.e., they can have combined effects when acting together. For instance, IL-3 and IL-4 amplify each other’s effects to induce growth, differentiation, and activation of mast cells in a synergistic manner [ 24 ]. Another phenomenon in cytokines signaling is antagonism. An example of cytokine antagonism is that cytokines of the IL-1 superfamily can antagonize IL-18 effects [ 54 ].
Cytokine categories according to immunological function
Four categories of cytokines are usually referred to in psychoimmunological literature. (1) T H 1 cytokines (IL-2, IL-12, IFN-γ) which induce cytotoxic cell contacts. (2) T H 2 cytokines (IL-4, (IL-5, IL-13) which lead to production of antibodies. (3) Pro-inflammatory cytokines (IL-1, IL-6, IL-8, IL-17, IL-21, IL-22, IFN-α, TNF-α) which further the progress of inflammation. (4) Anti-inflammatory cytokines (IL-10, transforming growth factor-beta (TGF)-β which are influenced by regulatory T cells and impede inflammatory process from escalating [ 24 ]. However, these categories are not distinct and it must be considered that cytokines can exert various effects on different cells and therefore, they may have pro- and also anti-inflammatory properties. For instance, IFN-α which has been listed as a pro-inflammatory cytokine can also have anti-inflammatory properties [ 55 ].
IL-6 as a pleiotropic cytokine
IL-6 was first identified as a differentiation factor for B cells which stimulates production of antibodies by activated B cells. Apart from regulation of acute inflammation, IL-6 is known to induce differentiation of B cells, and activation and population expansion of T cells [ 56 ]. Within the peripheral and central nervous system (CNS), IL-6 can act as a neuronal growth factor inducing neurite development and nerve regeneration [ 57 ]. IL-6 receptor (IL-6R) consists of the IL-6-binding chain which has two forms of transmembrane IL-6R and soluble IL-6R (sIL-6R) [ 58 ] and a gp130 signal-transducing chain [ 59 ]. Following binding to its receptor (IL-6R), IL-6 initiates to exert its multiple functions.
It is quite interesting that IL-6 exerts both pro- and anti-inflammatory properties [ 60 , 61 ]. Indeed, its signaling is complex and can lead to both inflammatory and anti-inflammatory cascades depending upon the presence of either IL-6 receptor (IL-6R) or the membrane bound gp130 signal transducer and these are expressed at very different frequencies within specific cell type in the body [ 5 ]. Trans-signaling of IL-6, in which the soluble form of the IL-6 receptor (sIL-6R) is shed from the membrane bound receptors, is known to be pro-inflammatory [ 62 ]. The sIL-6R binds to IL-6 and is transported to any cell type on which gp130 is expressed [ 63 ]. While most soluble receptors (e.g., soluble receptor for TNFα) result in antagonistic action by competing for the ligand, the sIL-6R is agonistic and increases the types of cells through which IL-6 can signal. Additionally, IL-6 engages in classical signaling which is anti-inflammatory [ 63 ] and occurs through binding of IL-6 to the membrane bound cell surface receptor. Classical signaling of IL-6 solely occurs on some subsets of T cells, neutrophils and monocytes megakaryocytes, and hepatocytes [ 64 ]. In both classical and trans-signaling, the IL-6/IL-6R/gp130 complex uses two pathways to activate intracellular signaling namely the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway and the mitogen-activated protein kinase (MAPK) pathway [ 5 ].
Indeed, IL-6 has been mostly regarded as having pro-inflammatory properties; however, it has many anti-inflammatory functions which are necessary for resolution of inflammation [ 65 ]. For instance, IL-6 inhibits activity of the transcription factor named nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and expression of the chemokine receptor on dendritic cells which is needed for recruiting these cells to lymphoid tissues; thus, involving in resolution of inflammation [ 66 ]. Research findings showed that IL-6 has a crucial role in regulation of T helper17 (Th17)/regulatory T (Treg) cells [ 67 ]. In the presence of TGF-β, IL-6 is a vital signal for differentiation of naive T cells into Th17 cells which in turn are implicated in induction of autoimmune diseases [ 68 , 69 ], and result in local tissue injury in chronic inflammatory disorders [ 70 ]. On the contrary, IL-6 can strongly inhibit the TGF-β-induced differentiation of Treg cells which in turn results in inhibition of autoimmunity and protects against tissue damage [ 71 ]. Functional dichotomy of IL-6 indicates that it may be responsible for maintaining the balance between pro- and anti-inflammatory responses, while having tissue-specific properties at the periphery and in the CNS [ 72 ].
Brief overview of chemokines and their role in depression
Chemokines are small chemotactic cytokines that are identified to have significant roles in migration of immune cells, induction of direct chemotaxis, and propagation of inflammatory responses [ 73 ]. They are classified into four sub-families based on their structural criteria (i.e., the number and spacing of their two N-terminals, disulfide bonding participating cysteine residues). These four subfamilies include CXC, CC, C, and CX3C [ 74 ]. Furthermore, they can be categorized according to their biological activity, namely, the maintenance of homeostasis and the induction of inflammation. There are also chemokines which have dual functionality [ 75 ].
These small chemotactic cytokines are known to be secreted in response to inflammatory cytokines; thereafter, selectively recruiting lymphocytes, monocytes, and neutrophil-inducing chemotaxis by activating G-protein-coupled receptors (GPCRs) [ 76 ]. A growing body of evidence suggests that chemotactic cytokines are implicated in neurobiological processes relevant to psychiatric disorders such as synaptic transmission and plasticity, neuroglia communication, and neurogenesis [ 77 ]. Indeed, disruption of any of the mentioned functions which may take place by activation of the inflammatory response system has consistently been found to be relevant in pathogenesis of depression [ 73 ].
There are indeed both pre-clinical and clinical evidence in support of linking alterations in the chemokine network to depressive behavior [ 73 ]. In an animal model of depression, namely prenatal stressed rats, levels of CCL2, and CXCL12 chemokines were found to be upregulated in the hippocampus and prefrontal cortex, which was indeed suggestive of excessive microglial activation [ 78 ]. Additionally, Trojan et al. (2017) investigated the modulatory properties of chronic administration of anti-depressants on the chemokines. According to their results, chronic administration of anti-depressants has been shown to normalize the prenatal stress-induced behavioral disturbances together with the observed alterations in CXCL12 and their receptor. Indeed, they concluded that alterations of CXCL12 and their receptor and at less extend changes in CX3CL1–CX3CR1 expression will probably be normalized following chronic treatment with anti-depressants [ 79 ].
Moreover, several clinical studies found correlations between elevated levels of circulating inflammatory chemokines and depressive symptoms in patients with major depressive disorder [ 73 , 80 , 81 , 82 , 83 ]. According to the results of a comprehensive meta-analysis, peripheral concentrations of a number of chemokines including CCL2, CCL3, CCL4, CCL11, CXCL4, CXCL7, and CXCL8 can potentially discriminate between individuals with depression and those without [ 84 ]. Additionally, Ślusarczyk et al. (2016) provided a comprehensive account of the role of chemokines in processes underlying depressive disorder [ 85 ].
Challenges of cytokine research in psychiatry
There are some difficulties faced by researchers in conducting cytokine research in psychiatry. The major problem seems to be heterogeneity of the obtained results. In other words, research outcomes are conflicting and challenging to interpret [ 24 ]. Moreover, research in this area is largely based on measurement of cytokine levels in the periphery and it is not completely clear how serum or plasma levels of cytokines reflect the situation in the brain [ 47 ]. Compellingly, results of studies that examined both peripheral and CFS levels of IL-6 found no correlations between the mentioned measures; thus, suggesting that peripheral levels of IL-6 may not directly reflect central IL-6 levels [ 86 , 87 ].
It is also noteworthy that some environmental, social, biological, and medical factors may influence peripheral cytokine changes. For instance, one of the characteristics of obesity is chronic inflammation with the increased circulating levels of cytokines [ 88 , 89 ]. Indeed, adipose tissue is reported to build up and activate lymphocytes and macrophages that secrete inflammatory factors [ 89 , 90 ]. Interestingly, obese people show behavioral symptoms such as MDD and cognitive dysfunction at an increased rate in comparison with the general population [ 91 , 92 ]. Therefore, one may argue that alterations in cytokine levels are somehow unspecific [ 24 ]. Additionally, there is a considerable overlap in cytokine values between patients in the acute phase of depression, patients in remission, and patients who are recovered [ 24 ]. Although the use of cytokines as potential biomarkers of depression has been discussed frequently in various studies, cytokine changes have been reported in other psychiatric disorders as well. For instance, increased levels of pro-inflammatory cytokines have been reported in generalized anxiety disorder [ 93 , 94 , 95 ], obsessive-compulsive disorder [ 96 ], posttraumatic stress disorder [ 97 , 98 ], and sleep disorder [ 99 ].
Moreover, cytokine levels change during pharmacotherapy of depression. Indeed, it has been suggested that treatment with anti-depressants can potentially lead to alteration in peripheral levels of cytokines. According to the results of a meta-analysis, anti-depressants, overall, cause decrease in peripheral levels of IL-6, IL-10, and TNF-α [ 100 ]. However, anti-psychotics which are used in psychotic depression, especially those with the highest risk of weight gain (e.g., clozapine and olanzapine), cause significant increase in the blood levels of pro-inflammatory cytokine [ 101 ]. Additionally, mood stabilizers such as lithium and carbamazepine have also been linked with an increase in the peripheral levels of cytokines [ 102 ]. In sum, as cytokine signaling often exhibits pleiotropic, redundant, synergistic, and antagonistic effects, it seems to be advisable to consider all cytokines that work together or against each other and therefore, take into account the whole range of cytokines instead of a single one.
On the role of interleukin-6 in depression
Il-6 alterations in depression.
A growing body of evidence suggests that IL-6 has a crucial role in pathogenesis of depression [ 3 ] and is the most consistently increased cytokine in blood samples of MDD patients [ 38 , 103 ]. The first promising evidence for the role of IL-6 in occurrence of depression is most probably provided by a longitudinal study in which children with higher circulating levels of IL-6 at age 9 were found to be at a 10% greater risk of developing MDD by age 18, compared to the general population or children with low IL-6 levels. Indeed, the researchers concluded that inflammation and high IL-6 levels possibly predate the occurrence of depression [ 104 ]. Another evidence for potential role of IL-6 in depression is that peripheral levels of IL-6 were found to be positively correlated with symptom severity in anti-depressant non-responders [ 105 ].
Stress-based preclinical models of depression showed that IL-6 levels are increased following the onset of depression-associated behaviors. Rodents who were exposed to chronic mild stress exhibited anhedonia and elevated circulating levels of pro-inflammatory cytokines including IL-6 [ 106 , 107 ]. Moreover, in another study on male Wistar rats, serum levels of pro-inflammatory cytokines including IL-6 were reported to be higher in acute and restraint stress compared to non-stressed rats [ 108 ]. It is also noteworthy that some studies reported no significant alteration in peripheral levels of IL-6 in chronic mild stress models of depression [ 109 ]. Nevertheless, they reported elevated CNS levels of other inflammatory markers which probably reflects a time-dependent shift from peripheral to central cytokine activation or potential transport of the peripheral cytokines into CNS [ 109 ]. Another promising evidence was provided by studies on IL-6 knockout mice. Indeed, they were reported to be resistant to the development of depression-like phenotype following long-term light deprivation in the constant darkness, proposing a functional role for IL-6 in stress susceptibility [ 110 ]. Moreover, Ślusarczyk et al. (2015) found evidence for the role of prenatal stress as a priming factor that could exhibit effects on microglial cells and consequently lead to depressive-like disturbances in adult rat offspring. According to their results, the release of pro-inflammatory cytokines including IL-6 is enhanced in microglia obtained from prenatally stressed animals compared to control animals [ 78 ].
In fact, not every individual exposed to prolonged or acute stress develops a psychiatric disorder [ 111 ]. According to previous research, vulnerability to repeated social defeat stress is predicted by differences in IL-6 levels in the innate peripheral immune system [ 112 ]. Following induction of social defeat stress, two thirds of mice were reported to show depression-like behavior measured by social avoidance, anhedonia, circadian system disruptions, and metabolic changes [ 113 ] together with elevated activation of pro-inflammatory cytokines such as IL-6 [ 112 ]. Indeed, higher degrees of elevation in peripheral IL-6 levels of susceptible mice were reported in comparison with resilient mice. Moreover, it was found that this increase occurs within 20 min of the first social defeat. Interestingly, mice that later became susceptible had higher number of leukocytes and those leukocytes produced more levels of IL-6 following stimulation via LPS ex vivo [ 112 ]. Additionally, studies with non-social stress-based models found evidence for the functional role of IL-6 in the development of stress susceptibility. In these models, animals were exposed to a controllable or uncontrollable stress (e.g., shock), and their ability to actively escape a subsequent stressor was measured. According to the results, 20% of animals who were exposed to uncontrollable stress were found susceptible and developed learned helplessness and the rest were found to be resilient. Interestingly, susceptible animals showed elevated levels of peripheral IL-6 together with anhedonia in contrast to resilient animals [ 114 ].
Clinical studies have also revealed that patients with MDD have increased levels of plasma and serum concentrations of pro-inflammatory cytokines including IL-6 in comparison with healthy controls [ 24 , 100 , 115 , 116 ]. It should be noted that three meta-analyses verified increased peripheral IL-6 levels in MDD patients compared to healthy volunteers [ 38 , 116 , 117 ]. Nevertheless, there are also studies reporting no significant differences in IL-6 levels in MDD patients compared to healthy volunteers [ 118 ]. However, one may argue that different subtypes of depression and certain depressive symptoms should be taken into account. For instance, Rudolf et al. (2014) compared IL-6 levels among patients with atypical and typical depression and healthy controls. According to their results, IL-6 levels were significantly increased in patients with atypical depression and not in typical MDD patients compared to healthy controls [ 119 ]. Additionally, Rush et al. (2016) studied peripheral levels of IL-6 and TGF-β in 55 melancholic depressed patients. They were found to have significantly higher baseline IL-6 levels compared to healthy controls. Moreover, these elevated levels of IL-6 did not normalize following electroconvulsive therapy (ECT) [ 120 ]. A recent systematic review conformed Rush et al.’s results. In the mentioned review, authors found that peripheral IL-6 levels are increased in patients with melancholic depression in comparison with controls [ 121 ]. Moreover, Maes et al. (1997) examined serum levels of IL-6 and IL-1 receptor antagonist in patients with chronic, treatment resistant depression both before and after subchronic treatment with anti-depressants. According to their results, subchronic treatment with anti-depressants had no significant impact on serum levels of IL-6; nevertheless, it decreased serum soluble IL-6R levels significantly [ 122 ].
Effects of IL-6 on neurotransmitters’ synthesis, signaling, metabolism, and function
The effects of cytokines on neurotransmitters have been studied extensively [ 49 , 123 ]. Cytokines and their signaling pathways (e.g., p38 mitogen activated protein kinase) are reported to exhibit significant impacts on metabolism of multiple neurotransmitters such as serotonin, dopamine, and glutamate; thus, influencing their synthesis, release, and reuptake [ 49 ]. Indeed, cytokines can decrease synthesis of serotonin via activating the enzyme indoleamine 2,3 dioxygenase (IDO) which breaks the precursor of serotonin (i.e., tryptophan) to kynurenine (KYN) instead of metabolizing tryptophan to serotonin; thus, leading to serotonin depletion [ 3 ]. The process of serotonin depletion has been long associated with major depression [ 124 , 125 ]. Moreover, cytokines can modulate serotonin signaling via elevating the expression and function of monoamine transporters. These transporters are known to re-uptake serotonin [ 126 , 127 ].
IL-6 is known to influence neurotransmission by modulating the behavioral output of the brain; however, the exact mechanism is unknown. A previous study showed that IL-6 directly controls the levels of serotonin transporter (SERT) and therefore influences serotonin reuptake. Indeed, the researchers concluded that IL6-induced modulation of serotonergic neurotransmission through the signal transducer and activator of transcription 3 (STAT3) signaling pathway contributes to the role of IL6 in depression [ 128 ]. The activity of SERT forms serotonergic transmission which is implicated in depressive behavioral changes and pathophysiology of the disease [ 129 ]. By intensifying dopaminergic and serotonergic turnover in hippocampus and frontal cortex, IL-6 influences neurotransmission of catecholamines [ 130 ]. It seems that noradrenaline is not affected by IL-6; however, noradrenaline itself can induce expression of IL-6 in glial cells [ 131 ]. IL-6 together with other pro-inflammatory cytokines can activate kinurenine pathway which is involved in glutamatergic neurotransmission [ 132 ].
Effects of IL-6 levels on brain morphology in depression
Previous studies showed that elevated levels of pro-inflammatory cytokines such as IL-6 may affect neurogenesis [ 133 ] and neural plasticity [ 134 ]. Imaging studies have shown that specific brain regions such as basal ganglia (which is involved in motor activity and motivation), the dorsal anterior cingulate cortex (ACC) (which has a central role in generation of anxiety), and the subgenual ACC (which is known to be involved in the development of depression) are influenced by cytokines [ 135 , 136 ]. Additionally, high IL-6 expression levels demonstrated neuropathologic manifestations including neurodegeneration [ 137 , 138 ].
There are many studies in which positron emission tomography (PET) has been applied to test translocator protein (TSPO) binding, a marker of neuroinflammation, in order to study neuroinflammatory hypothesis of depression [ 139 , 140 , 141 , 142 , 143 ]. According to their results, neuroinflammation was present in various regions of the brain (e.g., neocortical grey matter, frontal cortex, prefrontal cortex, anterior cingulate cortex, insula, temporal cortex) as well as the hippocampus [ 139 , 140 , 141 , 142 , 143 ].
In a recent study, Kakeda et al. (2018) evaluated possible relationship between serum levels of IL-1β, IL-6, IFN-γ, and TNFα and brain morphology in terms of brain cortical thinning and hippocampal subfield volumes during the first depressive episode in drug-naïve patients with MDD using a whole-brain SBM analysis. They found a significant inverse correlation between prefrontal cortex (PFC) thickness and serum IL-6 level in MDD patients. Indeed, high serum levels of IL-6 were correlated with reduced left subiculum and right CA1, CA3, CA4, GC-DG, subiculum, and whole hippocampus volumes in MDD patients. Additionally, thickness of the superior frontal and medial orbitofrontal cortices in patients with depression was significantly decreased compared to healthy controls. Since PFC contains high concentrations of IL-6 receptors, IL-6 mediated neurotoxicity might happen under conditions in which high serum IL-6 levels are present (i.e., early stages of MDD). Consequently, the authors advocated that the neuroinflammatory status in the early stage of MDD is associated with changes in the brain gray matter and IL-6 probably plays a key role in the morphological changes observed in the PFC during early stages of the disease. It is also noteworthy that serum IL-6 was the only cytokine among the tested cytokines that showed significant differences between the patients and controls in their study. Indeed, serum IL-6 levels were found to be significantly higher in MDD patients than in healthy controls [ 144 ]. In another study, Frodl et al. (2012) investigated possible effects of changes in the glucocorticoid and inflammatory systems on hippocampal volumes in patients with MDD. According to their results, MDD patients showed increased IL-6 levels and smaller hippocampal volumes compared to healthy controls. Positive effects of messenger RNA (mRNA) expression of glucocorticoid-inducible genes and further inverse effects of IL-6 concentration, on hippocampal volumes were also reported. Thus, they concluded that increased expression of IL-6 can probably predict decreased hippocampal volume [ 145 ].
As already mentioned, there is considerable amount of evidence regarding the central role of the highly plastic, stress-sensitive hippocampal region in pathogenesis of depression [ 146 ]. Indeed, grey-matter structures, including the hippocampus are vulnerable to atrophy in depression [ 147 , 148 ]. Hippocampal volume reductions are most probably the result of remodeling of key cellular elements, involving retraction of dendrites, loss of glial cells, and decreased neurogenesis in the dentate gyros [ 149 ]. Factors underlying this cellular remodeling are known to be stress-induced increased levels of glucocorticoids, which are implicated in decreased neurogenesis [ 150 ]. Moreover, increased activity of the HPA axis resulting in decreased levels of glucocorticoids combined with resistance to glucocorticoid-induced negative feedback control is commonly observed in depression [ 151 ]. This dysregulation of glucocorticoid secretion along with the increased activity of excitatory neurotransmitters can potentially lead to cellular remodeling (which can be reversible) and hippocampal neurons cell death in patients with depression [ 152 ]. Since hippocampus has been identified to have a role in negative feedback inhibition of glucocorticoids, remodeling or neuronal damage may lead to less efficient inhibitory control of the corticotrophin-releasing hormone, resulting in elevated amounts of circulating glucocorticoids and further damage of the hippocampal neurons [ 153 ]. Taken together, it seems that further studies are required to elucidate the physiological mechanisms in which IL-6 might exert changes in the brain grey matter. A brief overview of the effects of cytokines including IL-6 on brain morphology is shown in Fig. 1 .

Effects of cytokines including IL-6 on brain morphology
Blockade of IL-6 and its receptor in the periphery as a potential therapeutic option in MDD
Growing body of evidence suggests that abnormalities in the immune system are most probably relevant to pathogenesis and potential novel treatment of psychiatric disorders. Previous studies showed that alterations in the peripheral IL-6 levels might contribute to depressive-like behavior in animal studies [ 3 , 112 , 114 , 154 ]. Moreover, IL-6 knockout mice showed resistance to development of depressive-like behavior [ 155 ] which gives further evidence for possible role of IL-6 pathogenesis of depression. High peripheral levels of IL-6 are even more apparent in patients with treatment-resistant depression. Additionally, correlations have been found between decrease in IL-6 levels and alleviation of depressive symptoms in patients who were responsive to the pharmacotherapy [ 156 ]. Moreover, results of a study on 222 stroke patients indicated significant associations between IL-6 periphery levels and development of MDD within 2 weeks and at 1 year following stroke. Furthermore, significant correlations were found between statin use and IL-6 on the presence of a depressive disorder at the 1st year. Indeed, preventive effects of treatment with statins (which are known to possess anti-inflammatory properties and potently reduce the cytokine-mediated IL-6 release [ 157 ]) against post-stroke depression was confirmed [ 158 ]. Thus, suppression of IL-6 activity could possibly lead to clinical recovery and may be considered as a novel pharmacotherapeutic option. Utilizing IL-6 receptor antibodies (for instance, Tocilizumab) or IL-6 antibodies (for instance, Sirukumab or Siltuximab) for reduction of IL-6 activity seems to be a novel strategy.
Blockade of IL-6 receptor by the humanized anti-IL-6 antibody, Tocilizumab has been used in treatment of rheumatoid arthritis (RA) [ 159 , 160 , 161 , 162 , 163 ] and systemic juvenile idiopathic arthritis [ 164 , 165 , 166 ]. Extensive clinical studies have established both short-term and long-term efficacy and safety of Tocilizumab [±conventional disease-modifying anti-rheumatic drugs (DMARDs)] in adults with moderate to severe RA. Additionally, Tocilizumab was shown to be effective as a monotherapy in patients with systemic juvenile idiopathic arthritis and also in patients whose disease has been refractory to other therapies [ 164 ]. Moreover, the safety profile of tocilizumab was reported to be consistent over time and also consistent with safety profile of other immunomodulatory agents [ 162 ]. It is also important to note that oral tocilizumab has been shown to inhibit experimental autoimmune encephalitis by elevating Th2 anti-inflammatory cytokines and decreasing pro-inflammatory Th1 cytokines [ 167 ]. With regard to crucial role of IL-6 in regulating metabolic homeostasis, side effects such as significant weight gain followed by hypertrygliceridemia and hypercholesterolemia may be observed in patients treated with tocilizumab [ 168 ]. Blockade of IL-6 trans-signaling, while classical IL-6R signaling stays intact seems to be crucial for the goal of maintaining gut mucosal integrity and epithelial regeneration [ 65 ]. Indeed, few randomized clinical trials were conducted on anti-depressant properties of tocilizumab. According to the results of a recent meta-analysis of anti-depressant activity of anti-cytokine therapies, treatment with tocilizumab showed statistically significant improvements in depressive symptoms [ 169 ].
Another promising human monoclonal antibody against Il-6, namely Sirukumab has been reported to be a safe and well-tolerated agent, capable of modulating the immune response in healthy populations as well as in patients with inflammatory disorders (e.g., rheumatoid arthritis). It targets the IL-6 signaling pathway by inhibition of both the pro- and anti-inflammatory effects of IL-6 [ 170 ]. Effects of Sirukumab on cytokine networks provide a well-founded rationale for its potential use in pharmacotherapy of psychiatric disorders promising possible advantages across varying domains of the biobehavioral research criteria [ 171 ]. In a phase 2, double-blind, placebo-controlled trial, the efficacy of Sirukumab and Siltuximab on depressive symptoms was studied in patients with rheumatoid arthritis or multicentric Castleman’s disease respectively. Compared with placebo, both IL-6 neutralizing antibodies were found to make significantly greater improvements on depressive symptoms in the patients [ 172 ]. Results of a recent mega-analysis of 18 randomized, placebo-controlled clinical trials of efficacy of immunomodulatory drugs on depressive symptoms in patients with inflammatory disorders demonstrated promising results ( N = 10,743 participants). According to their findings, anti-IL-6 antibodies (sirukumab and siltuximab) had large and statistically significant effect sizes on core depressive symptoms before correction for physical health outcomes. Additionally, their effects remained significant in non-responders for the primary disease states evaluated [ 173 ]. Although further research is needed in this area, potential application of anti-IL-6 antibodies could possibly open new avenues in pharmacotherapy of MDD.
Possible role of IL-6 together with gut microbiota in pathogenesis of depression
The human intestine harbors nearly 100 trillion bacteria [ 174 ] consisting assemblages of microorganisms associated with various niches in and on the body with long-term implications to health [ 175 ]. Evidence is emerging regarding correlations of microbial activities with progressive structural and functional processes in the brain of both animal models and humans [ 175 ]. There is a large body of evidence for the role of gut microbiota composition in pathogenesis of depression [ 176 , 177 , 178 , 179 , 180 ]. Moreover, there is growing body of literature for the influence of the gut microbiome on cytokine signaling [ 181 , 182 ].
The dominant gut microbial phyla are known to be Firmicutes and Bacteroideteds [ 183 , 184 ]. The Firmicutes / Bacteroideteds ratio is of great relevance in signaling human gut microbiota status [ 185 ]. For instance, increased levels of Firmicutes / Bacteroideteds ratio have been reported in patients suffering from irritable bowel syndrome (IBD) and seem to have some correlations with development of depression and anxiety [ 186 , 187 ]. Additionally, Firmicutes / Bacteroideteds ratio is associated with overall alterations in bacterial profiles at different life stages [ 185 ]. In a novel study, researchers reported decreased Firmicutes / Bacteroideteds ratio in mice following social defeat stress; thus, proposing possible role of Firmicutes / Bacteroideteds in depressive-like behavior. Furthermore, administration of anti-mouse IL-6 receptor antibody (MR16-1) attenuated the decreased ratio of Firmicutes / Bacteroideteds in susceptible mice. Thus, the researchers concluded that anti-mouse IL-6 receptor antibody may have anti-depressant-like effects by normalizing the Firmicutes / Bacteroideteds ratio via modulation of the immune system [ 3 ].
Decreased number of Oscillospira was detected in patients with depression [ 180 ] which suggests for possible role of Oscillospira in pathogenesis of depression. Two animal studies yielded same results. A recent study investigated therapeutic effects of finasteride on depressive-like behavior in rats together with 1 month of treatment withdrawal. Withdrawal from finasteride was associated with increased depressive-like behavioral responses. Therapeutic use of finasteride was linked with elevations in the phylum Bacteroidetes and in the family Prevotellaceae, and withdrawal was found to be correlated with decreases in the family Ruminococcaceae and the genera Oscillospira and Lachnospira [ 188 ]. In another study, socially stressed mice developing depression-like symptoms showed increases at the genus level of fecal Oscillospira . Interestingly, IV administration of anti-mouse IL-6 receptor antibody (MR16-1) normalized depression-like behavior and resulted in significant decrease in Oscillospira levels towards pre-stressor levels [ 3 ]. Moreover, increased number of Sutterella was reported in fecal samples [ 189 ] and intestinal biopsy samples of children with Autism spectrum disorder [ 190 ]. Additionally, elevated number of Staphylococcus and Sutterella were found in mice following social defeat stress. It is likely that Staphylococcus and Sutterella play a role in the depressive-like behavior via infection-induced inflammation. Interestingly, administration of anti-mouse IL-6 receptor antibody resulted in attenuation of elevated number of Staphylococcus and Sutterella following social defeat stress in mice [ 3 ].
These findings advocate that peripheral IL-6 may have a significant role in pathogenesis of MDD and blockade of IL-6 receptor in the periphery may exhibit rapid-onset effects by attenuating the altered composition of gut microbiota. Taking into account the role of gut-microbiota in immunomodulation, it is highly probable that gut-microbiota-brain- axis plays a role in anti-depressant actions of treatment with anti-IL-6 receptor [ 3 ]. A brief overview of the role of IL-6 together with gut microbiota in pathogenesis of depression is shown in Fig. 2 .

A brief overview of the role of IL-6 together with gut microbiota in pathogenesis of depression
Elevated levels of IL-6 in patients with COVID-19 infection
The world-wide effect of the coronavirus disease 2019 (COVID-19) pandemic is enormous and is not solely limited to the increased mortality and morbidity rates, but also extends into the mental health of the global population [ 191 ]. Considerable amount of clinical data is emerging regarding the manifestation of depression in patients during [ 192 , 193 , 194 ] and post-COVID 19 infection [ 192 , 193 , 194 , 195 ]. It is estimated that about 48% of confirmed COVID-19 cases displayed overt psychological symptoms such as depression and often expressed feelings of regret, loneliness, helplessness, and irritation [ 196 ].
There is growing body of literature regarding dual role of IL-6 in COVID-19 infection and depression [ 192 ]. Normal plasma levels of IL-6 in adults range between 1 and 10 pg/ml; whereas in a systemic inflammation this amount increases to several ng/ml [ 197 ] and even higher concentrations were reported in COVID-19 patients [ 198 ]. Indeed, cytokine release syndrome (CRS) is common in COVID-19 patients and increased levels of serum IL-6 have been identified to be significantly associated with acute respiratory distress syndrome (ARDS), respiratory failure, and poor disease outcome in numerous studies [ 192 , 199 , 200 , 201 ]. Studies suggest that new onset depression is most probably caused by inflammation initiated during the active phase of the infection leading to a cytokine surge [ 202 ]. Indeed, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infects primarily human monocytes and dendritic cells causing dendritic cell dysfunction, leading to T cell apoptosis and exhaustion; thus, contributing to the immunopathology [ 203 ]. Alpert et al. (2020) described two cases of COVID-19 patients with elevated amounts of IL-6 (25 pg/mL and 26.7 pg/mL respectively) who were diagnosed with major depressive disorder (according to the Diagnostic and Statistical Manual of Mental Disorder, 5th Edition (DSM-5)) during COVID 19 infection. Both patients’ depressive symptoms subsided about 6 weeks after initiation of anti-depressant pharmacotherapy and normalization of the inflammatory cytokines [ 192 ]. The authors concluded that lower cytokine activity ameliorates depressive symptoms as normalization of IL-6 plasma levels decreased depression with or without anti-depressants [ 192 ]. Moreover, Benedett et al. (2020) studied effects of treatment with cytokine-blocking agents on the psychopathological status of the patients with COVID-19 infection. Their results were in favor of the protective effects of treatment with cytokine-blocking agents in early phases of COVID-19 against the later onset of depression [ 204 ].
It is indeed crucial to maintain a multidisciplinary approach in management of the psychological effects of this debilitating pandemic. Treatment strategies addressing the immunopathology of SARS-CoV-2 infection will be promising during the acute phase of the disease [ 192 , 205 ]. Currently, there are few studies considering psychological and neuropsychiatric implications of COVID-19; however, it is very likely to expect an increased incidence of mental pathologies both during and post-COVID-19 infection.
Preclinical and clinical studies present strong evidence that inflammation is altered in a subset of patients with MDD and there is mounting body of literature for the role of pro-inflammatory cytokines namely IL-6 in pathophysiology of depression. Nevertheless, there still exists gap in our understanding of the mechanisms by which IL-6 signaling and its molecular components could possibly contribute to depression manifestation. A number of humanized monoclonal antibodies are undergoing clinical trials for potential pharmacotherapy of mood disorders. Biologics including IL-6 receptor antibodies or IL-6 antibodies are currently approved to treat inflammatory disorders such as RA and are undergoing clinical trials as a novel target for MDD treatment. However, these novel therapeutic targets may also raise the possibility of potential side effects. By investigating the interface of peripheral cytokines, namely IL-6 and brain cellular processes contributing to depression, one might be able to develop novel therapeutic options for treatment of mood disorders by sequestering and preventing this peripherally derived inflammatory marker from acting upon mood circuits in the CNS. In sum, therapeutic deficiency in treatment outcomes reflects the growing demand for revitalizing psychiatric therapeutics with novel options that could potentially open new avenues in treatment of this debilitating disorder and enhancement of patients’ quality of life.
Availability of data and materials
Not applicable.
Abbreviations
Anterior cingulate cortex
Acute respiratory distress syndrome
Blood-brain barrier
Central nervous system
Coronavirus disease 2019
Cytokine release syndrome
Conserved transcriptional response to adversity
Disease-modifying anti-rheumatic drugs
Diagnostic and Statistical Manual of Mental Disorder, 5th Edition
Electroconvulsive therapy
Glycoprotein 130
G-protein-coupled receptors
Hypothalamic-pituitary-adrenal
Indoleamine 2,3 dioxygenase
Interleukin
IL-6 receptor
Janus kinase/signal transducer and activator of transcription
- Major depressive disorder
Mitogen-activated protein kinase
Messenger RNA
Nuclear factor kappa-light-chain-enhancer of activated B cells
Prefrontal cortex
Rheumatoid arthritis
severe acute respiratory syndrome coronavirus 2
Serotonin transporter
Signal transducer and activator of transcription 3
Soluble IL-6R
Transforming growth factor
T helper type 2
T helper type 1
T helper cell
Tumor necrosis factor
Translocator protein
Regulatory T cells
Depression and Other Common Mental Disorders: Global Health Estimates. 2017. Geneva: World Health Organization, Licence: CC BY-NC-SA 3.0 IGO.
Liu Q, He H, Yang J, Feng X, Zhao F, Lyu J. Changes in the global burden of depression from 1990 to 2017: Findings from the Global Burden of Disease study. J Psychiatr Res. 2020;126:134–40.
Article PubMed Google Scholar
Zhang JC, Yao W, Dong C, Yang C, Ren Q, Ma M, Hashimoto K. Blockade of interleukin-6 receptor in the periphery promotes rapid and sustained antidepressant actions: a possible role of gut-microbiota-brain axis. Transl Psychiatry. 2017;7:e1138.
Article CAS PubMed PubMed Central Google Scholar
Sim K, Lau WK, Sim J, Sum MY, Baldessarini RJ. Prevention of Relapse and Recurrence in Adults with Major Depressive Disorder: Systematic Review and Meta-Analyses of Controlled Trials. Int J Neuropsychopharmacol. 2016;19:pyv076.
Hodes GE, Ménard C, Russo SJ. Integrating Interleukin-6 into depression diagnosis and treatment. Neurobiol Stress. 2016;4:15–22.
Article PubMed PubMed Central Google Scholar
Fonseka TM, McIntyre RS, Soczynska JK, Kennedy SH. Novel investigational drugs targeting IL-6 signaling for the treatment of depression. Expert Opin Investig Drugs. 2015;24:459–75.
Article CAS PubMed Google Scholar
Miller AH, Raison CL. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol. 2016;16:22–34.
Majd M, Hashemian F, Hosseini SM, Vahdat Shariatpanahi M. Sharifi A. A randomized, double-blind, placebo-controlled trial of celecoxib augmentation of sertraline in treatment of drug-naive depressed women: a pilot study. Iran J Pharm Res. 2015;14:891–9.
CAS PubMed PubMed Central Google Scholar
Wohleb ES, Franklin T, Iwata M, Duman RS. Integrating neuroimmune systems in the neurobiology of depression. Nat Rev Neurosci. 2016;17:497–511.
Zhao G, Liu X. Neuroimmune advance in depressive disorder. Adv Exp Med Biol. 2019;1180:85–98.
Lee CH, Giuliani F. The role of inflammation in depression and fatigue. Front Immunol. 2019;10:1696.
Liu CH, Zhang GZ, Li B, Li M, Woelfer M, Walter M, Wang L. Role of inflammation in depression relapse. J Neuroinflammation. 2019;16:90.
Cassano T, Calcagnini S, Carbone A, Bukke VN, Orkisz S, Villani R, Romano A, Avolio C, Gaetani S. Pharmacological treatment of depression in Alzheimer’s disease: a challenging task. Front Pharmacol. 2019;10:1067.
Hirschfield RM. History and evolution of the monoamine hypothesis of depression. J Clin Psychiatry. 2000;61:4–6.
Google Scholar
Massart R, Mongeau R, Lanfumey L. Beyond the monoaminergic hypothesis: neuroplasticity and epigenetic changes in a transgenic mouse model of depression. PhilosTrans R Soc Lond Ser B Biol Sci. 2012;367:2485–94.
Article CAS Google Scholar
Boku S, Nakagawa S, Toda H, Hishimoto A. Neural basis of major depressive disorder: Beyond monoamine hypothesis. Psychiatry Clin Neurosci. 2018;72:3–12.
Kopschina Feltes P, Doorduin J, Klein HC, Juárez-Orozco LE, Dierckx RA. Moriguchi- Jeckel CM, de Vries EF. Anti-inflammatory treatment for major depressive disorder: implications for patients with an elevated immune profile and non-responders to standard antidepressant therapy. J Psychopharmacol. 2017;31:1149–65.
Maes M, Van der Planken M, Stevens WJ, Peeters D, DeClerck LS, Bridts CH, Schotte C, Cosyns P. Leukocytosis, monocytosis and neutrophilia: hallmarks of severe depression. J Psychiatr Res. 1992;26:125–134.
Maes M, Lambrechts J, Bosmans E, Jacobs J, Suy E, Vandervorst C, de Jonckheere C, Minner B, Raus J. Evidence for a systemic immune activation during depression: results of leukocyte enumeration by flow cytometry in conjunction with monoclonal antibody staining. Psychol Med. 1992;22:45–53.
Maes M, Scharpé S, Meltzer HY, Bosmans E, Suy E, Calabrese J, Cosyns P. Relationships between interleukin-6 activity, acute phase proteins, and function of the hypothalamic- pituitary-adrenal axis in severe depression. Psychiatry Res. 1993;49:11–27.
Slavich GM, Irwin MR. From stress to inflammation and major depressive disorder: a social signal transduction theory of depression. Psychol Bull. 2014;140:774–815.
Irwin MR, Miller AH. Depressive disorders and immunity: 20 years of progress and discovery. Brain Behav Immun. 2007;21:374–83.
Henter ID, de Sousa RT, Gold PW, Brunon AR, Zarate CA, Machado-Vieira R. Mood therapeutics: novel pharmacological approaches for treating depression. Expert Rev Clin Pharmacol. 2017;10:153-166.
Himmerich H, Patsalos O, Lichtblau N, Ibrahim MAA, Dalton B. Cytokine research in depression: principles, challenges, and open questions. Front Psychiatry. 2019;7(10):30.
Article Google Scholar
Zou W, Feng R, Yang Y. Changes in the serum levels of inflammatory cytokines in antidepressant drug-naïve patients with major depression. PLoS One. 2018;13:e0197267.
Article PubMed PubMed Central CAS Google Scholar
Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatr. 2009;65:732–41.
Horowitz MA, Zunszain PA. Neuroimmune and neuroendocrine abnormalities in depression: two sides of the same coin. Ann N Y Acad Sci. 2015;1351:68–79.
Raison CL. Miller AH. Is depression an inflammatory disorder? Curr Psychiatry Rep. 2011;13:467–75.
Young JJ, Bruno D. Pomara N. A review of the relationship between proinflammatory cytokines and major depressive disorder. J Affect Disord. 2014;169:15–20.
Kim YK, Na KS, Shin KH, Jung HY, Choi SH, Kim JB. Cytokine imbalance in the pathophysiology of major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1044–53.
Himmerich H, Kohls E, Hegerl U, Rummel-Kluge C. Prädiktive Faktoren der Depression und ihrer Therapie. Der Nervenarzt. 2014;85:1249–54.
Muñoz RF, Cuijpers P, Smit F, Barrera AZ, Leykin Y. Prevention of major depression. Ann Rev Clin Psychol. 2010;6:181–212.
Brebner K, Hayley S, Zacharko R, Merali Z, Anisman H. Synergistic effects of interleukin-1β, interleukin-6, and tumor necrosis factor-α: Central monoamine, corticosterone, and behavioral variations. Neuropsychopharmacology. 2000;22:566–80.
Farooq RK, Asghar K, Kanwal S, Zulqernain A. Role of inflammatory cytokines in depression: focus on interleukin-1β. Biomed Rep. 2017;6:15–20.
Felger JC, Lotrich FE. Inflammatory cytokines in depression: neurobiological mechanisms and therapeutic implications. Neuroscience. 2013;246:199–229.
Black C, Miller BJ. Meta-analysis of cytokines and chemokines in suicidality: distinguishing suicidal versus nonsuicidal patients. Biol Psychiatry. 2015;78:28–37.
Goldsmith DR, Rapaport MH. Miller BJ. A meta-analysis of blood cytokine network alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder and depression. Mol Psychiatry. 2016;21:1696–709.
Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK. Lanctôt KL. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67:446–57.
Strawbridge R, Arnone D, Danese A, Papadopoulos A, Herane Vives A, Cleare AJ. Inflammation and clinical response to treatment in depression: A meta-analysis. Eur Neuropsychopharmacol. 2015;25:1532–43.
Fan N, Luo Y, Ou Y, He H. Altered serum levels of TNF-α, IL-6, and IL-18 in depressive disorder patient. Hum Psychopharmacol. 2017;32(4) https://doi.org/10.1002/hup.2588 .
Nishuty NL, Khandoker MH, Karmoker JR, Ferdous S, Shahriar M, Qusar S, Islam S. Kadir MF, Islam R. Evaluation of serum interleukin-6 and C-reactive protein levels in drug-naïve major depressive disorder patient. cureus. 2019;11:e3868.
PubMed PubMed Central Google Scholar
Ramesh G, MacLean AG, Philipp MT. Cytokines and chemokines at the crossroads of neuroinflammation, neurodegeneration. and neuropathic pain. Mediators Inflamm. 2013;2013:480739.
PubMed Google Scholar
Arango Duque G, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol. 2014;7(5):491.
Galic MA, Riazi K, Pittman QJ. Cytokines and brain excitability. Front Neuroendocrinol. 2012;33:116–25.
Wilson CJ, Finch CE, Cohen HJ. Cytokines and cognition--the case for a head-to-toe inflammatory paradigm. J Am Geriatr Soc. 2002;50:2041–56.
Hopkins SJ. Central nervous system recognition of peripheral inflammation: a neural. hormonal collaboration. Acta Biomed. 2007;78:231–47.
Capuron L, Miller AH. Immune system to brain signaling: neuropsychopharmacological implications. Pharmacol Ther. 2011;130:226–38.
Caldwell AB, Cheng Z, Vargas JD, Birnbaum HA, Hoffmann A. Network dynamics determine the autocrine and paracrine signaling functions of TNF. Genes Dev. 2014;28:2120–33.
Miller AH, Haroon E, Raison CL, Felger JC. Cytokine targets in the brain: impact on neurotransmitters and neurocircuits. Depress Anxiety. 2013;30:297–306.
Perskidskii IV, Barshtein IA. Biological manifestations of the tumor necrosis factor effect and its role in the pathogenesis of various diseases. Arkh Patol. 1992;54:5–10.
Zhang JM, An J. Cytokines, inflammation. and pain. Int Anesthesiol Clin. 2007;45:27–37.
Kicielinska J, Pajtasz-Piasecka E. The role of IL-10 in the modulation of the immune response in normal conditions and the tumor environment. Postepy Higieny Med Doświadczal. 2014;68:879–92.
Munk ME, Emoto M. Functions of T-cell subsets and cytokines in mycobacterial infections. Eur Respir J Suppl. 1995;20:668s–−675s.
CAS PubMed Google Scholar
Krumm B, Xiang Y, Deng J. Structural biology of the IL-1 superfamily: key cytokines in the regulation of immune and inflammatory responses. Protein Sci. 2014;23:526–38.
Tilg H, Peschel C. Interferon-alpha and its effects on the cytokine cascade: a pro- and anti-inflammatory cytokine. Leukem Lymphoma. 1996;23:55–60.
Klimpel GR. Soluble factor(s) from LPS-activated macrophages induce cytotoxic T cell differentiation from alloantigen-primed spleen cells. J Immunol. 1980;125:1243–9.
Spooren A, Kolmus K, Laureys G, Clinckers R, De Keyser J, Haegeman G, Gerlo S. Interleukin-6. a mental cytokine. Brain Res Rev. 2011;67:157–83.
Yamasaki K, Taga T, Hirata Y, Yawata H, Kawanishi Y, Seed B, Taniguchi T, Hirano T, Kishimoto T. Cloning and expression of the human interleukin-6 (BSF-2/IFN beta 2) receptor. Science. 1988;241:825–8.
Hibi M, Murakami M, Saito M, Hirano T, Taga T, Kishimoto T. Molecular cloning and expression of an IL-6 signal transducer, gp130. Cell. 1990;63:1149–57.
Fuster JJ, Walsh K. The good, the bad, and the ugly of interleukin-6 signaling. EMBO J. 2014;33:1425–7.
Schett G. Physiological effects of modulating the interleukin-6 axis. Rheumatology. 2018;57:ii43–ii50.
Mullberg J, Schooltink H, Stoyan T, Gunther M, Graeve L, Buse G, Mackiewicz A. Heinrich PC, Rose-John S. The soluble interleukin-6 receptor is generated by shedding. Eur J Immunol. 1993;23:473–80.
Wolf J, Rose-John S, Garbers C. Interleukin-6 and its receptors: a highly regulated and dynamic system. Cytokine. 2014;70:11–20.
Scheller J, Chalaris A, Schmidt-Arras D, Rose-John S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim Biophys Acta. 2011;1813:878–88.
Hunter CA, Jones SA. IL-6 as a keystone cytokine in health and disease. Nat Immunol. 2015;16:448–57.
Hegde S, Pahne J, Smola-Hess S. Novel immunosuppressive properties of interleukin- 6 in dendritic cells: inhibition of NF-kB binding activity and CCR7 expression. FASEB J. 2004;18:1439–41.
Kimura A, Kishimoto T. IL-6: regulator of Treg/Th17 balance. Eur J Immunol. 2010;40:1830–5.
Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–4.
Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–8.
Iwakura Y, Ishigame H, Saijo S, Nakae S. Functional specialization of interleukin-17 family members. Immunity. 2011;34:149–62.
Dominitzki S, Fantini MC, Neufert C, Nikolaev A, Galle PR, Scheller J, Monteleone G, Rose-John S, Neurath MF, Becker C. Cutting edge: trans-signaling via the soluble IL-6R abrogates the induction of FoxP3 in naïve CD4+CD25 T cells. J Immunol. 2007;179:2041–5.
Milovan BM, Ivan J, Gordana R, Jelena P, Slavica MJ, Nebojsa A, Lukic ML. Interleukin-6 in schizophrenia—is there a therapeutic relevance? Front Psychiatry. 2017;8:221.
Milenkovic VM, Stanton EH, Nothdurfter C, Rupprecht R, Wetzel CH. The role of chemokines in the pathophysiology of major depressive disorder. Int J Mol Sci. 2019;20:2283.
Article CAS PubMed Central Google Scholar
Zlotnik A, Yoshie O. The chemokine superfamily revisited. Immunity. 2012;36:705–16.
Le Thuc O, Blondeau N, Nahon JL, Rovère C. The complex contribution of chemokines to neuroinflammation: switching from beneficial to detrimental effects. Ann N Y Acad Sci. 2015;1351:127–40.
Article PubMed CAS Google Scholar
Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res. 2009;29:313–26.
Stuart MJ, Baune BT. Chemokines and chemokine receptors in mood disorders, schizophrenia, and cognitive impairment: a systematic review of biomarker studies. Neurosci Biobehav Rev. 2014;42:93–115.
Ślusarczyk J, Trojan E, Głombik K, Budziszewska B, Kubera M, Lasoń W, Popiołek- Barczyk K, Mika J, Wędzony K, Basta-Kaim A. Prenatal stress is a vulnerability factor for altered morphology and biological activity of microglia cells. Front Cell Neurosci. 2015;9:82.
Trojan E, Ślusarczyk J, Chamera K, Kotarska K, Głombik K, Kubera M, Basta-Kaim A. The modulatory properties of chronic antidepressant drugs treatment on the brain chemokine – chemokine receptor network: a molecular study in an animal model of depression. Front Pharmacol. 2017;8:779.
Simon NM, McNamara K, Chow CW, Maser RS, Papakostas GI, Pollack MH, Nierenberg AA, Fava M. Wong KK. A detailed examination of cytokine abnormalities in major depressive disorder. Eur Neuropsychopharmacol. 2008;18:230–3.
Piletz JE, Halaris A, Iqbal O, Hoppensteadt D, Fareed J, Zhu H, Sinacore J, Devane CL. Pro- inflammatory biomakers in depression: treatment with venlafaxine. World J Biol Psychiatry. 2009;10:313–23.
De la Peña FR, Cruz-Fuentes C, Palacios L, Girón-Pérez MI, Medina-Rivero E. Ponce- Regalado MD, Alvarez-Herrera S, Pérez-Sánchez G, Becerril-Villanueva E, Maldonado-García JL, Jiménez-Martínez MC, Pavón L. Serum levels of chemokines in adolescents with major depression treated with fluoxetine. World J Psychiatry. 2020;10:175–86.
Romero-Sanchiz P, Nogueira-Arjona R, Araos P, Serrano A, Barrios V, Argente J, Garcia- Marchena N, Lopez-Tellez A, Rodriguez-Moreno S, Mayoral F, et al. Variation in chemokines plasma concentrations in primary care depressed patients associated with Internet-based cognitive-behavioral therapy. Sci Rep. 2020;10:1078.
Leighton SP, Nerurkar L, Krishnadas R, Johnman C, Graham GJ, Cavanagh J. Chemokines in depression in health and in inflammatory illness: a systematic review and meta- analysis. Mol Psychiatry. 2018;23:48–58.
Ślusarczyk J, Trojan E, Chwastek J, Głombik K, Basta-Kaim AA. Potential Contribution of Chemokine Network Dysfunction to the Depressive Disorders. Curr Neuropharmacol. 2016;14:705–20.
Sasayama D, Hattori K, Wakabayashi C, Teraishi T, Hori H, Ota M, Yoshida S, Arima K, Higuchi T, Amano N, et al. Increased cerebrospinal fluid interleukin-6 levels in patients with schizophrenia and those with major depressive disorder. J Psychiatr Res. 2013;47:401–6.
Boufidou F, Lambrinoudaki I, Argeitis J, Zervas IM, Pliatsika P, Leonardou AA. Petropoulos G, Hasiakos D, Papadias K, Nikolaou C. CSF and plasma cytokines at delivery and postpartum mood disturbances. J Affect Disord. 2009;115:287–92.
Schmidt FM, Weschenfelder J, Sander C, Minkwitz J, Thormann J, Chittka T, Mergl R, Kirkby KC, Faßhauer M, Stumvoll M, et al. Inflammatory cytokines in general and central obesity and modulating effects of physical activity. PLoS ONE. 2015;10:e0121971.
Bastard JP, Jardel C, Delattre J, Hainque B, Bruckert E, Oberlin F. Evidence for a link between adipose tissue interleukin-6 content and serum C-reactive protein concentrations in obese subjects. Circulation. 1999;99:2221–2.
Wellen KE, Hotamisligil GS. Obesity-induced inflammatory changes in adipose tissue. J Clin Invest. 2003112:1785–8.
Sweat V, Starr V, Bruehl H, Arentoft A, Tirsi A, Javier E. Convit A. C-reactive protein is linked to lower cognitive performance in overweight and obese women. Inflammation. 2008;31:198–207.
Capuron L, Su S, Miller AH, Bremner JD, Goldberg J, Vogt GJ, Maisano C, Jones L. Murrah NV, Vaccarino V. Depressive symptoms and metabolic syndrome: is inflammation the underlying link? Biol Psychiatry. 2008;64:896–900.
Hou R, Garner M, Holmes C, Osmond C, Teeling J, Lau L, Baldwin DS. Peripheral inflammatory cytokines and immune balance in Generalised Anxiety Disorder: Case- controlled study. Brain Behav Immun. 2017;62:212–8.
Costello H, Gould RL, Abrol E, Howard R. Systematic review and meta-analysis of the association between peripheral inflammatory cytokines and generalised anxiety disorder. BMJ Open. 2019;9:e027925.
Vieira MMM, Ferreira TB, Pacheco PAF, Barros PO, Almeida CRM, Araújo-Lima CF, Silva-Filho RG, Hygino J, Andrade RM, Linhares UC, et al. Enhanced Th17 phenotype in individuals with generalized anxiety disorder. J Neuroimmunol. 2010;229:212–8.
Gray SM, Bloch MH. Systematic review of proinflammatory cytokines in obsessive- compulsive disorder. Curr Psychiatr Rep. 2012;14:220–8.
Hori H, Kim Y. Inflammation and post-traumatic stress disorder. PCN Psychiatry and Clinical Neurosciences. 2019;73:143–53.
Renna ME, O’Toole MS, Spaeth PE, Lekander M, Mennin DS. The association between anxiety, traumatic stress, and obsessive-compulsive disorders and chronic inflammation: a systematic review and meta-analysis. Depress Anxiety. 2018;35:1081–94.
Irwin MR. Why sleep is important for health: a psychoneuroimmunology perspective. Annu Rev Psychol. 2015;66:143–72.
Köhler CA, Freitas TH, Stubbs B, Maes M, Solmi M, Veronese N, de Andrade NQ, Morris G, Fernandes BS, Brunoni AR, et al. Peripheral alterations in cytokine and chemokine levels after antidepressant drug treatment for major depressive disorder: systematic review and meta-analysis. Mol Neurobiol. 2018;55:4195–206.
Himmerich H, Minkwitz J, Kirkby K. Weight gain and metabolic changes during treatment with antipsychotics and antidepressants. Endocr Metabol Immune Disorder Drug Targets. 2015;15:252–60.
Himmerich H, Koethe D, Schuld A, Yassouridis A, Pollmacher T. Plasma levels of leptin and endogenous immune modulators during treatment with carbamazepine or lithium. Psychopharmacology. 2005;179:447–51.
Haapakoski R, Mathieu J, Ebmeier KP, Alenius H, Kivimaki M. Cumulative meta- analysis of interleukins 6 and 1beta, tumour necrosis factor alpha and C-reactive protein in patients with major depressive disorder. Brain Behav Immun. 2015;49:206–15.
Khandaker GM, Pearson RM, Zammit S, Lewis G, Jones PB. Association of serum interleukin 6 and C-reactive protein in childhood with depression and psychosis in young adult life: a population-based longitudinal study. JAMA Psychiatry. 2014;71:1121–8.
Lanquillon S, Krieg JC, Bening-Abu-Shach U, Vedder H. Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology. 2000;22:370–9.
Mutlu O, Gumuslu E, Ulak G, Celikyurt IK, Kokturk S, Kir HM, Akar F, Erden F. Effects of fluoxetine, tianeptine and olanzapine on unpredictable chronic mild stress-induced depression-like behavior in mice. Life Sci. 2012;91:1252–62.
Pan Y, Zhang WY, Xia X, Kong LD. Effects of icariin on hypothalamic-pituitary- adrenal axis action and cytokine levels in stressed Sprague-Dawley rats. Biol Pharm Bull. 2006;29:2399–403.
Himmerich H, Fischer J, Bauer K, Kirkby KC, Sack U, Krugel U. Stress-induced cytokine changes in rats. Eur Cytokine Network. 2013;24:97–103.
Farooq RK, Isingrini E, Tanti A, Le Guisquet AM, Arlicot N, Minier F, Leman S, Chalon S, Belzung C. Camus V. Is unpredictable chronic mild stress (UCMS) a reliable model to study depression-induced neuroinflammation? Behav Brain Res. 2012;231:130–7.
Monje FJ, Cabatic M, Divisch I, Kim EJ, Herkner KR, Binder BR, Pollak DD. Constant darkness induces IL-6-dependent depression-like behavior through the NF-kappaB signaling pathway. J Neurosci. 2011;31:9075–83.
Russo SJ, Murrough JW, Han MH, Charney DS, Nestler EJ. Neurobiology of resilience. Nat Neurosci. 2012;15:1475–84.
Hodes GE, Pfau ML, Leboeuf M, Golden SA, Christoffel DJ, Bregman D, Rebusi N, Heshmati M, Aleyasin H, Warren BL, et al. Individual differences in the peripheral immune system promote resilience versus susceptibility to social stress. Proc Natl Acad Sci U S A. 2014;111:16136–41.
Krishnan V. Molecular adaptations underlying susceptibility and resistance to social defeat in brain reward regions. Cell. 2007;131:391–404.
Yang C, Shirayama Y, Zhang JC, Ren Q, Hashimoto K. Peripheral interleukin-6 promotes resilience versus susceptibility to inescapable electric stress. Acta Neuropsychiatr. 2015;27:312–6.
Wang M, Wei J, Yang X, Ni P, Wang Y, Zhao L, Deng W, Guo W, Wang Q, Li T, et al. The level of IL-6 was associated with sleep disturbances in patients with major depressive disorder. Neuropsychiatric Disease and Treatment. 2019;15:1695–700.
Howren MB, Lamkin DM, Suls J. Associations of depression with C-reactive protein, IL-1, and IL-6: a meta-analysis. Psychosom Med. 2009;71:171–86.
Liu Y, Ho RC, Mak A. Interleukin (il)-6, tumour necrosis factor alpha (tnf-alpha) and soluble interleukin-2 receptors (sil-2r) are elevated in patients with major depressive disorder: A meta-analysis and meta-regression. J Affect Disord. 2012;139:230–9.
Chocano-Bedoya PO, Mirzaei F, O’Reilly EJ, Lucas M, Okereke OI, Hu FB, Rimm EB, Ascherio A. C-reactive protein, interleukin-6, soluble tumor necrosis factor alpha receptor 2 and incident clinical depression. J Affect Disord. 2014;163:25–32.
Rudolf S, Greggersen W, Kahl KG, Huppe M, Schweiger U. Elevated il-6 levels in patients with atypical depression but not in patients with typical depression. Psychiatry Res. 2014;217:34–8.
Rush G, O’Donovan A, Nagle L, Conway C, McCrohan A, O’Farrelly C, Lucey JV, Malone KM. Alteration of immune markers in a group of melancholic depressed patients and their response to electroconvulsive therapy. J Affect Disord. 2016;205:60–8.
Yang C, Tiemessen KM, Bosker FJ, Wardenaar KJ, Lie J, Schoevers RA. Interleukin, tumor necrosis factor-alpha and c-reactive protein profiles in melancholic and non-melancholic depression: A systematic review. J Psychosom Res. 2018;111:58–68.
Maes M, Bosmans E, De Jongh R, Kenis G, Vandoolaeghe E, Neels H. Increased serum IL- 6 and IL-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine. 1997;9:853–8.
Lichtblau N, Schmidt FM, Schumann R, Kirkby KC, Himmerich H. Cytokines as biomarkers in depressive disorder: current standing and prospects. Int Rev Psychiatry. 2013;25:592–603.
Cowen PJ, Browning M. What has serotonin to do with depression? World Psychiatry. 2015;14:158–60.
Albert PR, Benkelfat C, Descarries L. The neurobiology of depression—revisiting the serotonin hypothesis. I. Cellular and molecular mechanisms. Philos Trans R Soc Lond B Biol Sci. 2012;367:2378–81.
Zhu CB, Carneiro AM, Dostmann WR, Hewlett WA. Blakely RD. p38 MAPK Activation Elevates Serotonin Transport Activity via a Trafficking-independent, Protein Phosphatase 2A-dependent Proces. The Journal of Biological Chemistry. 2005;280:15649–58.
Zhu CB, Blakely RD, Hewlett WA. The proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha activate serotonin transporters. Neuropsychopharmacology. 2006;31:2121–31.
Kong E, Sucic S, Monje FJ, Reisinger SN, Savalli G, Diao W, Khan D, Ronovsky M, Cabatic M, Koban F, et al. STAT3 controls IL6-dependent regulation of serotonin transporter function and depression-like behavior. Sci Rep. 2015;5:9009.
Baudry A, Pietri M, Launay JM, Kellermann O, Schneider B. Multifaceted regulations of the serotonin transporter: impact on antidepressant response. Front Neurosci. 2019;13:91.
Zalcman S, Green-Johnson JM, Murray L, Nance DM, Dyck D, Anisman H, Greenberg AH. Cytokine-specific central monoamine alterations induced by interleukin-1, -2 and – 6. Brain Res. 1994;643:40–9.
Day JS, O’Neill E, Cawley C, Aretz NK, Kilroy D, Gibney SM, Harkin A, Conor TJ. Noradrenaline acting on astrocyticβ2-adrenoceptors induces neurite outgrowth in primary cortical neurons. Neuropharmacology. 2014;77:234–48.
Müller N, Myint AM, Krause D, Weidinger E, Schwarz MJ. Anti-inflammatory treatment in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2013;42:146–53.
Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–5.
Khairova RA, Machado-Vieira R, Du J. Manji HK. A potential role for pro-inflammatory cytokines in regulating synaptic plasticity in major depressive disorder. Int J Neuropsychopharmacol. 2009;12:561–78.
Alexander GE, Crutcher MD, DeLong MR. Basal ganglia-thalamocortical circuits: parallel substrates for motor, oculomotor, “prefrontal” and “limbic” functions. Progress Brain Res. 1991;85:119–46.
Eisenberger NI, Lieberman MD. Why rejection hurts: a common neural alarm system for physical and social pain. Trends Cogn Sci. 2004;8:294–300.
Rothaug M, Becker-Pauly C, Rose-John S. The role of interleukin-6 signaling in nervous tissue. Biochim Biophys Acta. 2016;1863:1218–27.
Campbell IL, Abraham CR, Masliah E, Kemper P, Inglis JD, Oldstone MB, Mucke L. Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc Natl Acad Sci U S A. 1993;90:10061–5.
Setiawan E, Attwells S, Wilson AA, Mizrahi R, Rusjan PM, Miler L, Xu C, Sharma S, Kish S, Houle S, et al. Association of translocator protein total distribution volume with duration of untreated major depressive disorder: a cross-sectional study. The Lancet Psychiatry. 2018;5:339–47.
Setiawan E, Wilson AA, Mizrahi R, Rusjan PM, Miler L, Rajkowska G, Suridjan I, Kennedy JL, Rekkas PV, Houle S, et al. Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry. 2015;72:268–75.
Holmes SE, Hinz R, Conen S, Gregory CJ, Matthews JC, Anton-Rodriguez JM, Gerhard A, Talbot PS. Elevated translocator protein in anterior cingulate in major depression and a role for inflammation in suicidal thinking: a positron emission tomography study. Biol Psychiatry. 2018;83:61–9.
Richards EM, Zanotti-Fregonara P, Fujita M, Newman L, Farmer C, Ballard ED, Machado- Vieira R, Yuan P, Niciu MJ, Lyoo CH, et al. PET radioligand binding to translocator protein (TSPO) is increased in unmedicated depressed subjects. EJNMMI Res. 2018;8:57.
Li H, Sagar AP, Kéri S. Translocator protein (18kDa TSPO) binding, a marker of microglia, is reduced in major depression during cognitive-behavioral therapy. Prog Neuropsychopharmacol Biol Psychiatry. 2018;83:1–7.
Kakeda S, Watanabe K, Katsuki A, Sugimoto K, Igata N, Ueda I, Igata R, Abe O, Yoshimura R, Korogi Y. Relationship between interleukin (IL)-6 and brain morphology in drug-naïve, first-episode major depressive disorder using surface-based morphometry. Sci Rep. 2018;8:10054.
Frodl T, Carballedo A, Hughes MM, Saleh K, Fagan A, Skokauskas N, McLoughlin DM, Meaney J, O'Keane V, Connor TJ. Reduced expression of glucocorticoid-inducible genes GILZ and SGK-1: high IL-6 levels are associated with reduced hippocampal volumes in major depressive disorder. Transl Psychiatry. 2012;2:e88.
Campbell S, Macqueen G. The role of the hippocampus in the pathophysiology of major depression. J Psychiatry Neurosci. 2004;29:417–26.
MacQueen GM, Campbell S, McEwen BS, Macdonald K, Amano S, Joffe RT, Nahmias C, Young LT. Course of illness, hippocampal function, and hippocampal volume in major depression. Proc Natl Acad Sci U S A. 2003;100:1387-1392.
Frodl T, Meisenzahl EM, Zetzsche T. Born C, Groll C, Jäger M, Leinsinger G, Bottlender R, Hahn K, Möller HJ. Hippocampal changes in patients with a first episode of major Depression. Am J Psychiatry. 2002;159:1112–8.
Rajkowska G. Postmortem studies in mood disorders indicate altered numbers of neurons and glial cells. Biol Psychiatry. 2000;48:766–7.
Cameron HA, McKay RD. Restoring production of hippocampal neurons in old age. Nat Neurosci. 1999;2:894–7.
Young EA, Haskett RF, Grunhaus L, Pande A, Weinberg VM, Watson SJ, Akil H. Increased evening activation of the hypothalamic-pituitary-adrenal axis in depressed patients. Arch Gen Psychiatry. 1994;51:701–7.
Sapolsky RM. The possibility of neurotoxicity in the hippocampus in major depression: a primer on neuron death. Biol Psychiatry. 2000 Oct 15;48(8):755–65.
McEwen BS. The neurobiology of stress: from serendipity to clinical relevance. Brain Res. 2000;886:172–89.
Yang C, Hashimoto K. Peripheral IL-6 signaling: a promising therapeutic target for depression? Expert Opin Investig Drugs. 2015;24:989–90.
Chourbaji S, Urani A, Inta I, Sanchis-Segura C, Brandwein C, Zink M, Schwaninger M, Gass P. IL-6 knockout mice exhibit resistance to stress-induced development of depression-like behaviors. Neurobiol Dis. 2006;23:587–94.
Ting EYC, Yang AC, Tsai AJ. Role of interleukin-6 in depressive disorder. Int J Mol Sci. 2020;21:2194.
Loppnow H, Zhang L, Buerke M, Lautenschläger M, Chen L, Frister A, Schlitt A, Luther T, Song N, Hofmann B, et al. Statins potently reduce the cytokine-mediated IL-6 release in SMC/MNC cocultures. J Cell Mol Med. 2011;15:994–1004.
Kang HJ, Bae KY, Kim SW, Kim JT, Park MS, Cho KH, Kim JM. Effects of interleukin-6, interleukin-18, and statin use, evaluated at acute stroke, on post-stroke depression during 1- year follow-up. Psychoneuroendocrinology. 2016;72:156–60.
Burmester GR, Rigby WF, van Vollenhoven RF, Kay J, Rubbert-Roth A, Kelman A, Dimonaco S, Mitchell N. Tocilizumab in early progressive rheumatoid arthritis: FUNCTION, a randomised controlled trial. Ann Rheum Di. 2016;75:1081-1091.
Yazici Y, Curtis JR, Ince A, Baraf H, Malamet RL, Teng LL, Kavanaugh A. Efficacy of tocilizumab in patients with moderate to severe active rheumatoid arthritis and a previous inadequate response to disease-modifying antirheumatic drugs: the ROSE study. Ann Rheum Dis. 2012;71:198–205.
Kaneko A. Tocilizumab in rheumatoid arthritis: efficacy, safety and its place in therapy. Ther Adv Chronic Dis. 2013;4:15–21.
Scott LJ. Tocilizumab: a review in rheumatoid arthritis. Drugs. 2017;77:1865–79.
Haraoui B, Casado G, Czirják L, Taylor A, Dong L, Button P, Luder Y, Caporali R. Tocilizumab patterns of use, effectiveness, and safety in patients with rheumatoid arthritis: final results from a set of multi-national non-interventional studies. Rheumatol Ther. 2019;6:231–43.
Turnier JL, Brunner HI. Tocilizumab for treating juvenile idiopathic arthritis. Expert Opin Biol Ther. 2016;16:559–66.
Machado SH, Xavier RM. Safety of tocilizumab in the treatment of juvenile idiopathic arthritis. Expert Opin Drug Saf. 2017;16:493–500.
Shepherd J, Cooper K, Harris P, Picot J, Rose M. The clinical effectiveness and cost- effectiveness of abatacept, adalimumab, etanercept and tocilizumab for treating juvenile idiopathic arthritis: a systematic review and economic evaluation. Health Technol Assess. 2016;20:1–222.
Brod SA, Bauer VL. Ingested (oral) tocilizumab inhibits EAE. Cytokine. 2014;68:86–93.
Nishimoto N, Kanakura Y, Aozasa K, Johkoh T, Nakamura M, Nakano S, Nakano N, Ikeda Y, Sasaki T, Nishioka K, et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood. 2005;106:2627–32.
Kappelmann N, Lewis G, Dantzer R, Jones PB, Khandaker GM. Antidepressant activity of anti-cytokine treatment: a systematic review and meta-analysis of clinical trials of chronic inflammatory conditions. Mol Psychiatry. 2018;23:335–43.
Taylor PC, Schiff MH, Wang Q, Jiang Y, Zhuang Y, Kurrasch R, Daga S, Rao R, Tak PP, Hsu B. Efficacy and safety of monotherapy with sirukumab compared with adalimumab monotherapy in biologic-naïve patients with active rheumatoid arthritis (SIRROUND-H): a randomised, double-blind, parallel-group, multinational, 52-week, phase 3 study. Annals of the Rheumatic Diseases. 2018;77:658–66.
Zhou AJ, Lee Y, Salvadore G, Hsu B, Fonseka TM, Kennedy SH, McIntyre RS. Sirukumab: a potential treatment for mood disorders? Adv Ther. 2017;34:78–90.
Sun Y, Wang D, Salvadore G, Hsu B, Curran M, Casper C, Vermeulen J, Kent JM, Singh J, Drevets WC, et al. The effects of interleukin-6 neutralizing antibodies on symptoms of depressed mood and anhedonia in patients with rheumatoid arthritis and multicentric castleman’s disease. Brain Behav. Immun. 2017;66:156–64.
Wittenberg GM, Stylianou A, Zhang Y, Sun Y, Gupta A, Jagannatha PS, Wang D, Hsu B, Curran ME, Khan S, et al. Effects of immunomodulatory drugs on depressive symptoms: A mega-analysis of randomized, placebo-controlled clinical trials in inflammatory disorders. Mol Psychiatry. 2020;25:1275–85.
Guinane CM, Cotter PD. Role of the gut microbiota in health and chronic gastrointestinal disease: understanding a hidden metabolic organ. Therap Adv Gastroenterol. 2013;6:295–308.
Sharon G, Sampson TR, Geschwind DH, Mazmanian SK. The central nervous system and the gut microbiome. Cell. 2016;167:915–32.
Cheung SG, Goldenthal AR, Uhlemann AC, Mann JJ, Miller JM, Sublette ME. Systematic review of gut microbiota and major depression. Front Psychiatry. 2019;10:34.
Kelly JR, Clarke G, Cryan JF, Dinan TG. Brain-gut-microbiota axis: challenges for translation in psychiatry. Ann Epidemiol. 2016;26:366–72.
Fung T, Olson C, Hsiao E. Interactions between the microbiota, immune and nervous systems in health and disease. Nat Neurosci. 2017;20:145–55.
Jiang H, Ling Z, Zhang Y, Mao H, Ma Z, Yin Y, Wang W, Tang W, Tan Z, Shi J, Li L, Ruan B. Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav Immun. 2015;48:186–94.
Zheng P, Zeng B, Zhou C, Liu M, Fang Z, Xu X, Zeng L, Chen J, Fan S. Du X. Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host's metabolism. Mol Psychiatry. 2016;21:786–96.
Schirmer M, Smeekens SP, Vlamakis H, Jaeger M, Oosting M, Franzosa EA, Ter Horst R, Jansen T, Jacobs L, Bonder MJ, et al. Linking the human gut microbiome to inflammatory cytokine production capacity. Cell. 2016;167:1125–36 e8.
O'Mahony L, McCarthy J, Kelly P, Hurley G, Luo F, Chen K, O'Sullivan GC, Kiely B, Collins JK, Shanahan F, et al. Lactobacillus and bifidobacterium in irritable bowel syndrome: symptom responses and relationship to cytokine profiles. Gastroenterology. 2005;128:541–51.
Rinninella E, Raoul P, Cintoni M, Franceschi F, Miggiano GAD, Gasbarrini A, Mele MV. What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet. and diseases. Microorganisms. 2019;7:14.
Lloyd-Price J, Abu-Ali G, Huttenhower C. The healthy human microbiome. Genome Med. 2016;8:51.
Mariat D, Firmesse O, Levenez F, Guimarăes V, Sokol H, Doré J, Corthier G, Furet JP. The Firmicutes/Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 2009;9:123.
Jeffery IB, O’Toole PW, Öhman L, Claesson MJ, Deane J, Quigley EM, Simrén M. An irritable bowel syndrome subtype defined by species-specific alterations in faecal microbiota. Gut. 2012;61:997–1006.
Distrutti E, Monaldi L, Ricci P, Fiorucci S. Gut microbiota role in irritable bowel syndrome: New therapeutic strategies. World J Gastroenterol. 2016;22:2219–41.
Diviccaro S, Giatti S, Borgo F, Barcella M, Borghi E, Trejo JL, Garcia-Segura LM, Melcangi RC. Treatment of male rats with finasteride, an inhibitor of 5alpha-reductase enzyme, induces long-lasting effects on depressive-like behavior, hippocampal neurogenesis, neuroinflammation and gut microbiota composition. Psychoneuroendocrinology. 2019;99:206–15.
Wang L, Christophersen CT, Sorich MJ, Gerber JP, Angley MT, Conlon MA. Increased abundance of Sutterella spp. and Ruminococcus torques in feces of children with autism spectrum disorder. Mol Autism. 2013;4:42.
Williams BL, Hornig M, Parekh T, Lipkin WI. Application of novel PCR-based methods for detection, quantitation, and phylogenetic characterization of Sutterella species in intestinal biopsy samples from children with autism and gastrointestinal disturbances. MBio. 2012;3:pii: e00261–11.
Guo Q, Zheng Y, Shi J, Wang J, Li G, Li C. Immediate psychological distress in quarantined patients with COVID-19 and its association with peripheral inflammation: a mixed-method study. Brain Behav Immun. 2020;88:17–27.
Alpert O, Begun L, Garren P, Solhkhah R. Cytokine storm induced new onset depression in patients with COVID-19. A new look into the association between depression and cytokines -two case reports. Brain Behav Immun Health. 2020;9:100173.
Taquet M, Luciana S, Geddes JR, Harrison PJ. Bidirectional associations between COVID- 19 and psychiatric disorder: retrospective cohort studies of 62354 COVID-19 cases in the USA. Lancet Psychiatry. 2020. https://doi.org/10.1016/S2215-0366(20)30462-4 .
Mukaetova-Ladinska, EB, Kronenberg G. Psychological and neuropsychiatric implications of COVID-19. Eur Arch Psychiatry Clin Neurosci. 2020. https://doi.org/10.1007/s00406 - 020-01210-2
Mazza MG, De Lorenzo R, Conte C, Poletti S, Vai B, Bollettini I, Melloni EMT, Furlan R, Ciceri F, Rovere-Querini P, et al. Anxiety and depression in COVID-19 survivors: Role of inflammatory and clinical predictors. Brain Behav Immun. 2020;89:594–600.
Ahmed MZ, Ahmed O, Aibao Z, Hanbin S, Siyu L, Ahmad A. Epidemic of COVID-19 in China and associated psychological problems. Asian J Psychiatr. 2020;51:10209.
Steardo L, Steardo L, Verkhratsky A. Psychiatric face of COVID-19. Transl Psychiatry. 2020;10:261.
Coomes EA, Haghbayan H. Interleukin-6 in Covid-19: a systematic review and meta- analysis. Rev Med Virol. 2020;30:1–9.
Mojtabavi H, Saghazadeh A, Rezaei N. Interleukin-6 and severe COVID-19: a systematic review and meta-analysis. Eur Cytokine Netw. 2020;31:44–9.
Sun H, Guo P, Zhang L, Wang F. Serum Interleukin-6 Concentrations and the Severity of COVID-19 Pneumonia: A Retrospective Study at a Single Center in Bengbu City, Anhui Province, China. in January and February 2020. Med Sci Monit. 2020;26:e926941.
Gubernatorova EO, Gorshkova EA, Polinova AI, Drutskaya MS. IL-6: relevance for immunopathology of SARS-CoV-2. Cytokine Growth Factor Rev. 2020;53:13–24.
Liu B, Li M, Zhou Z, Guan X, Xiang Y. Can we use interleukin-6 (IL-6) blockade for coronavirus disease 2019 (COVID-19)-induced cytokine release syndrome (CRS)? J Autoimmun. 2020:102452.
Moore JB, June CH. Cytokine release syndrome in severe COVID- 19. Science. 2020;368:473–4.
Benedetti F, Mazza M, Cavalli G, Ciceri F, Dagna L, Rovere-Querini P. Can cytokine blocking prevent depression in COVID-19 survivors? J Neuroimmune Pharmacol. 2020:1–3.
Catanzaro M, Fagiani F, Racchi M, Corsini E, Govoni S, Lanni C. Immune response in COVID-19: addressing a pharmacological challenge by targeting pathways triggered by SARS-CoV-2. Sig Transduct Target Ther. 2020;5:84.
Download references
Acknowledgements
Not applicable
Financial support was solely provided by the authors.
Author information
Authors and affiliations.
Department of Clinical Pharmacy, Faculty of Pharmacy, Tehran Medical Sciences, Islamic Azad University, No. 99 Yakhchal Street, Shariati Avenue, Tehran, 1941933111, Iran
Elnaz Roohi & Farshad Hashemian
Université de Poitiers, Unité de recherche clinique intersectorielle Pierre Deniker du Centre Hospitalier Henri Laborit F-86022 France, Groupement De Recherche CNRS 3557, Poitiers, France
Nematollah Jaafari
You can also search for this author in PubMed Google Scholar
Contributions
ER contributed to the conception of the study, searched, screened, did the data extraction of the studies, drafted, and reviewed the manuscript. NJ contributed to the conception of the study, reviewed, and edited the manuscript. FH conceived the idea of the study, critically reviewed, and edited the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Correspondence to Farshad Hashemian .
Ethics declarations
Ethics approval and consent to participate, consent for publication, competing interests.
The authors declare that they have no competing interests.
Additional information
Publisher’s note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ . The Creative Commons Public Domain Dedication waiver ( http://creativecommons.org/publicdomain/zero/1.0/ ) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
Reprints and permissions
About this article
Cite this article.
Roohi, E., Jaafari, N. & Hashemian, F. On inflammatory hypothesis of depression: what is the role of IL-6 in the middle of the chaos?. J Neuroinflammation 18 , 45 (2021). https://doi.org/10.1186/s12974-021-02100-7
Download citation
Received : 09 November 2020
Accepted : 02 February 2021
Published : 16 February 2021
DOI : https://doi.org/10.1186/s12974-021-02100-7
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Neuroinflammation
- Interleukin-6
Journal of Neuroinflammation
ISSN: 1742-2094
- Submission enquiries: [email protected]
- Get Press Releases

- Media Contacts
- News Releases
- Photos & B-Roll Downloads
- VUMC Facts and Figures
Explore by Highlight
- Education & Training
- Growth & Finance
- Leadership Perspectives
- VUMC People
Explore by Topic
- Emergency & Trauma
- Genetics & Genomics
- Health Equity
- Health Policy
- Patient Spotlight
- Tech & Health
Explore by Location
- Monroe Carell Jr. Children’s Hospital at Vanderbilt
- Vanderbilt Bedford County Hospital
- Vanderbilt Health One Hundred Oaks
- Vanderbilt Health Affiliated Network
- Vanderbilt-Ingram Cancer Center
- Vanderbilt Psychiatric Hospital
- Vanderbilt Wilson County Hospital
- Vanderbilt Stallworth Rehabilitation Hospital
- Vanderbilt Tullahoma Harton Hospital
- Vanderbilt University Hospital

- Photos & B-Roll Downloads
Featured Story

VUMC performs its first combined lung and liver transplant
April 9, 2024, heart disease, depression linked by inflammation: study.
Coronary artery disease and major depression may be genetically linked via inflammatory pathways to an increased risk for cardiomyopathy, a degenerative heart muscle disease, researchers at Vanderbilt University Medical Center and Massachusetts General Hospital have found.

Their report, published April 5 in the journal Nature Mental Health , suggests that drugs prescribed for coronary artery disease and depression, when used in combination, potentially may reduce inflammation and prevent the development of cardiomyopathy.

“This work suggests that chronic low-level inflammation may be a significant contributor to both depression and cardiovascular disease,” said the paper’s corresponding author, Lea Davis , PhD, associate professor of Medicine in the Division of Genetic Medicine and Vanderbilt Genetics Institute.
The connection between depression and other serious health conditions is well known. As many as 44% of patients with coronary artery disease (CAD), the most common form of cardiovascular disease, also have a diagnosis of major depression. Yet the biological relationship between the two conditions remains poorly understood.
A possible connection is inflammation. Changes in the levels of inflammatory markers have been observed in both conditions, suggesting that there may be a common biological pathway linking neuroinflammation in depression with atherosclerotic inflammation in CAD.
In the current study, the researchers used a technique called transcriptome-wide association scans to map single nucleotide polymorphisms (genetic variations) involved in regulating the expression of genes associated with both CAD and depression.
The technique identified 185 genes that were significantly associated with both depression and CAD, and which were “enriched” for biological roles in inflammation and cardiomyopathy. This suggests that predisposition to both depression and CAD, which the researchers called (major) depressive CAD, or (m)dCAD, may further predispose individuals to cardiomyopathy.
However, when the researchers scanned large electronic health record databases at VUMC, Mass General, and the National Institutes of Health’s All of Us Research Program, they found the actual incidence of cardiomyopathy in patients with the enriched genes for (m)dCAD was lower than in patients with CAD alone.
One possible explanation is that medications prescribed for CAD and depression, such as statins and antidepressants, may prevent development of cardiomyopathy by reducing inflammation, the researchers concluded.
“More research is needed to investigate optimal treatment mechanisms,” Davis added, “but at a minimum this work suggests that patient heart and brain health should be considered together when developing management plans to treat depression or cardiovascular disease.”
Kritika Singh, PhD, the paper’s first author, is a former graduate student in the Davis lab who is now a postdoctoral Innovation Fellow at Novartis in Cambridge, Massachusetts.
Other VUMC co-authors are Tyne Miller-Fleming, PhD, Peter Straub, MS, Nancy Cox, PhD, founding director of the Vanderbilt Genetics Institute, and institute members Quinn Wells, MD, PharmD, MSCI, associate professor of Medicine in the Division of the Cardiovascular Medicine, and Emily Hodges, PhD, assistant professor of Biochemistry.
The research was supported by National Institutes of Health grants R56MH120736, R01H118233, 1F31MH124306, and 1R01HL140074, and an American Heart Association Fellowship.
Related Articles

February 15, 2023
Study finds chronically disrupted sleep may increase risk for heart disease.
Vanderbilt research found that sleep irregularity — chronically disrupted sleep and highly variable sleep durations night after night — may increase the risk for atherosclerosis.
By Bill Snyder

December 18, 2014
Atrial disease and hypertension links.
New findings suggest that misfolded proteins accumulate in the heart atria, particularly in patients with hypertension, and may contribute to atrial heart disease.
By Leigh Macmillan

February 2, 2023
Researchers clarify role of blood cell mutations in disease.
Vanderbilt researchers have developed a new method to analyze mutations in blood stem cells that can trigger explosive, clonal expansions of abnormal cells.
- Alzheimer's disease & dementia
- Arthritis & Rheumatism
- Attention deficit disorders
- Autism spectrum disorders
- Biomedical technology
- Diseases, Conditions, Syndromes
- Endocrinology & Metabolism
- Gastroenterology
- Gerontology & Geriatrics
- Health informatics
- Inflammatory disorders
- Medical economics
- Medical research
- Medications
- Neuroscience
- Obstetrics & gynaecology
- Oncology & Cancer
- Ophthalmology
- Overweight & Obesity
- Parkinson's & Movement disorders
- Psychology & Psychiatry
- Radiology & Imaging
- Sleep disorders
- Sports medicine & Kinesiology
- Vaccination
- Breast cancer
- Cardiovascular disease
- Chronic obstructive pulmonary disease
- Colon cancer
- Coronary artery disease
- Heart attack
- Heart disease
- High blood pressure
- Kidney disease
- Lung cancer
- Multiple sclerosis
- Myocardial infarction
- Ovarian cancer
- Post traumatic stress disorder
- Rheumatoid arthritis
- Schizophrenia
- Skin cancer
- Type 2 diabetes
- Full List »
share this!
April 8, 2024
This article has been reviewed according to Science X's editorial process and policies . Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
Heart disease and depression may be genetically linked by inflammation
by Vanderbilt University Medical Center
. DOI: 10.1038/s44220-024-00219-z")
Coronary artery disease and major depression may be genetically linked via inflammatory pathways to an increased risk for cardiomyopathy, a degenerative heart muscle disease, researchers at Vanderbilt University Medical Center and Massachusetts General Hospital have found.
Their report, published April 5 in the journal Nature Mental Health , suggests that drugs prescribed for coronary artery disease and depression, when used in combination, potentially may reduce inflammation and prevent the development of cardiomyopathy.
"This work suggests that chronic low-level inflammation may be a significant contributor to both depression and cardiovascular disease," said the paper's corresponding author, Lea Davis, Ph.D., associate professor of Medicine in the Division of Genetic Medicine and Vanderbilt Genetics Institute.
The connection between depression and other serious health conditions is well known. As many as 44% of patients with coronary artery disease (CAD), the most common form of cardiovascular disease, also have a diagnosis of major depression . Yet the biological relationship between the two conditions remains poorly understood.
A possible connection is inflammation. Changes in the levels of inflammatory markers have been observed in both conditions, suggesting that there may be a common biological pathway linking neuroinflammation in depression with atherosclerotic inflammation in CAD.
In the current study, the researchers used a technique called transcriptome-wide association scans to map single nucleotide polymorphisms (genetic variations) involved in regulating the expression of genes associated with both CAD and depression.
The technique identified 185 genes that were significantly associated with both depression and CAD, and which were "enriched" for biological roles in inflammation and cardiomyopathy. This suggests that predisposition to both depression and CAD, which the researchers called (major) depressive CAD, or (m)dCAD, may further predispose individuals to cardiomyopathy.
However, when the researchers scanned large electronic health record databases at VUMC, Mass General, and the National Institutes of Health's All of Us Research Program, they found the actual incidence of cardiomyopathy in patients with the enriched genes for (m)dCAD was lower than in patients with CAD alone.
One possible explanation is that medications prescribed for CAD and depression, such as statins and antidepressants, may prevent development of cardiomyopathy by reducing inflammation, the researchers concluded.
"More research is needed to investigate optimal treatment mechanisms," Davis added, "but at a minimum this work suggests that patient heart and brain health should be considered together when developing management plans to treat depression or cardiovascular disease."
Kritika Singh, Ph.D., the paper's first author, is a former graduate student in the Davis lab who is now a postdoctoral Innovation Fellow at Novartis in Cambridge, Massachusetts.
Explore further
Feedback to editors

Two key brain systems are central to psychosis, study finds
3 hours ago

COVID-19 vaccine effectiveness: Results from Norway demonstrate the reproducibility of federated analytics
7 hours ago

Elucidating the link between Guillain–Barré syndrome and Takotsubo cardiomyopathy
8 hours ago

Artificial intelligence can help people feel heard, study finds

A new diagnostic model offers hope for Alzheimer's
9 hours ago

New study validates prediction rules for pediatric intra-abdominal and traumatic brain injuries

Chemicals stored in home garages linked to amyotrophic lateral sclerosis risk

In the drive to deprescribe, heartburn drug study teaches key lessons

Researchers identify new genetic risk factors for persistent HPV infections

New AI method captures uncertainty in medical images
Related stories.

Using cardiac MRI to investigate cause of cardiomyopathy in coronary artery disease
Nov 10, 2023

For younger women, mental health now may predict heart health later
Mar 28, 2024

Study links depression scores, white blood cell count
Dec 3, 2021

Prenatal depression may be linked to cardiovascular disease after childbirth
Apr 19, 2023

Depression linked to heart disease and type 2 diabetes
Feb 16, 2022

Inflammation links heart disease and depression, study finds
Mar 18, 2019
Recommended for you

Scientists use wearable technology to detect stress levels during sleep

Survey of mental health and exposure to blasts reveals differences among displaced people who remained in Ukraine

Study unpicks why childhood maltreatment continues to impact on mental and physical health into adulthood
10 hours ago

Researchers identify safety of a potential new treatment to manage complications from sickle cell disease
Let us know if there is a problem with our content.
Use this form if you have come across a typo, inaccuracy or would like to send an edit request for the content on this page. For general inquiries, please use our contact form . For general feedback, use the public comments section below (please adhere to guidelines ).
Please select the most appropriate category to facilitate processing of your request
Thank you for taking time to provide your feedback to the editors.
Your feedback is important to us. However, we do not guarantee individual replies due to the high volume of messages.
E-mail the story
Your email address is used only to let the recipient know who sent the email. Neither your address nor the recipient's address will be used for any other purpose. The information you enter will appear in your e-mail message and is not retained by Medical Xpress in any form.
Newsletter sign up
Get weekly and/or daily updates delivered to your inbox. You can unsubscribe at any time and we'll never share your details to third parties.
More information Privacy policy
Donate and enjoy an ad-free experience
We keep our content available to everyone. Consider supporting Science X's mission by getting a premium account.
E-mail newsletter
- Open access
- Published: 09 April 2024
Association of major depression, schizophrenia and bipolar disorder with thyroid cancer: a bidirectional two-sample mendelian randomized study
- Rongliang Qiu 1 , 2 ,
- Huihui Lin 3 ,
- Hongzhan Jiang 3 ,
- Jiali Shen 3 ,
- Jiaxi He 4 &
- Jinbo Fu 1 , 2
BMC Psychiatry volume 24 , Article number: 261 ( 2024 ) Cite this article
96 Accesses
2 Altmetric
Metrics details
Major depressive disease (MDD), schizophrenia (SCZ), and bipolar disorder (BD) are common psychiatric disorders, and their relationship with thyroid cancer has been of great interest. This study aimed to investigate the potential causal effects of MDD, SCZ, BD, and thyroid cancer.
We used publicly available summary statistics from large-scale genome-wide association studies to select genetic variant loci associated with MDD, SCZ, BD, and thyroid cancer as instrumental variables (IVs), which were quality controlled and clustered. Additionally, we used three Mendelian randomization (MR) methods, inverse variance weighted (IVW), MR–Egger regression and weighted median estimator (WME) methods, to estimate the bidirectional causal relationship between psychiatric disorders and thyroid cancer. In addition, we performed heterogeneity and multivariate tests to verify the validity of the IVs.
We used two-sample bidirectional MR analysis to determine whether there was a positive causal association between MDD and thyroid cancer risk. The results of the IVW analysis (OR = 3.956 95% CI = 1.177–13.299; P = 0.026) and the WME method (OR = 5.563 95% CI = 0.998–31.008; P = 0.050) confirmed that MDD may increase the risk of thyroid cancer. Additionally, our study revealed a correlation between genetic susceptibility to SCZ and thyroid cancer (OR = 1.532 95% CI = 1.123–2.088; P = 0.007). The results of the WME method analysis based on the median estimate (OR = 1.599 95% CI = 1.014–2.521; P = 0.043) also suggested that SCZ may increase the risk of thyroid cancer. Furthermore, our study did not find a causal relationship between BD and thyroid cancer incidence. In addition, the results of reverse MR analysis showed no significant causal relationships between thyroid cancer and MDD, SCZ, or BD ( P > 0.05), ruling out the possibility of reverse causality.
Conclusions
This MR method analysis provides new evidence that MDD and SCZ may be positively associated with thyroid cancer risk while also revealing a correlation between BD and thyroid cancer. These results may have important implications for public health policy and clinical practice. Future studies will help elucidate the biological mechanisms of these associations and potential confounders.
Peer Review reports
Cancer is recognized as a disease that poses a serious threat to human health, and it has been reported that in the United States alone, more than 609,360 people are expected to lose their lives to cancer in 2022 [ 1 ]. However, the mechanisms underlying the development of most cancers are still not fully understood, which has led to delays in the diagnosis and treatment of cancers, contributing to the increasing incidence and mortality of cancer worldwide. Thyroid cancer is one of the most common endocrine tumours, and its incidence has been steadily increasing worldwide over the last three decades due to the widespread use of diagnostic imaging techniques and ultrasound-guided fine needle aspiration (US-FNA) [ 2 , 3 ]. Despite the continued increase in the incidence of thyroid cancer, its mortality trend has remained relatively stable. Risk factors for thyroid cancer include metabolic syndrome (including diabetes mellitus, hypertension, obesity, etc.), poor lifestyle habits, and environmental pollution, but these factors do not fully explain the mechanism of thyroid cancer development. Therefore, identifying other potentially modifiable risk factors, such as psychiatric disorders, is important for the prevention and treatment of thyroid cancer.
Major depressive disease (MDD), schizophrenia (SCZ), and bipolar disorder (BD) are all serious psychiatric disorders that overlap genetically and clinically, suggesting that they may share common aetiological mechanisms [ 4 ]. The results of a study suggest that quantitative changes in plasma lipids affect several individual characteristics, including those affected by serious psychiatric disorders (MDD, SCZ and BD) [ 5 ]. Moreover, clinical studies have shown that patients with MDD, SCZ and BD have altered Homer1a levels in specific regions and cell types of the brain. A growing body of research confirms the close connection between these three disorders [ 6 ]. MDD is ranked by the World Health Organization as one of the most burdensome diseases in the world; it seriously damages people’s physical and mental health and is associated with a variety of endocrine disorders, such as hypothyroidism and hyperthyroidism [ 7 , 8 , 9 , 10 , 11 ]. Studies have shown that patients with hyperthyroidism and hypothyroidism differ in the presentation of depressive symptoms and disorders [ 12 , 13 ]. Specifically, hyperthyroidism was associated with more depressive symptoms (e.g., insomnia and weight loss) [ 14 ], whereas hypothyroidism was associated with fewer depressive symptoms (e.g., energy deficit and fatigue) [ 15 ]. In addition, individuals with hyperthyroidism have a higher incidence of MDD [ 13 ]. The relationship between depression and cancer has long been of interest, and some observational studies have suggested that depression may be an important risk factor for cancer [ 16 ]. A cross-sectional study from Korea revealed a 5.6% incidence of depression in thyroid cancer patients [ 17 ]. Another study from Germany showed that cancer patients were five times more likely to be depressed than was the general population, and thyroid cancer patients with a detectable high burden of depressive symptoms were 9.3 times more likely to be depressed than was the general population [ 18 ]. However, despite observational studies revealing a correlation between MDD and thyroid cancer, the relationship between MDD and thyroid cancer has not been systematically explored.
SCZ is a chronic psychiatric disorder accompanied by inconsistent behavioural and cognitive symptoms and has profound effects on both individuals and society. More than 50% of those diagnosed have intermittent and chronic psychiatric problems [ 19 ]. This results in a particularly high risk of disengagement from the labour market, with employment rates ranging from 10 to 30%, unemployment rates as high as 89–95%, and a 15–20 year reduction in life expectancy [ 20 , 21 ]. There is growing evidence that thyroid function may be altered in patients with SCZ, but the results of observational studies have been inconsistent [ 22 , 23 ]. In addition, the role of the thyroid gland in the pathophysiology of SCZ is poorly understood, and the relationship between thyroid disorders and SCZ is unclear.
SCZ and BD are considered part of the psychiatric continuum and share similar clinical features. BD is a chronic, disabling illness and a major contributor to the global burden of disease. BD can cause mood swings ranging from depression to mania. Patients exhibit fluctuations during the course of the illness, with some patients experiencing episodes only every few years, while others experience episodes almost continuously. A large body of evidence confirms the association between abnormal thyroid hormone levels and different psychopathological conditions, triggering neuropsychiatric symptoms [ 24 ]. However, observational studies may find an association between psychiatric disorders and thyroid disorders, but confounding factors and reverse causality cannot be excluded.
To explore the causal association between psychiatric disorders (MDD, SCZ and BD) and thyroid cancer risk, we used a two-sample bidirectional Mendelian randomization (MR) study. MR studies [ 25 ] use genetic variation as an instrumental variable closely related to the exposure of interest to explore the causal effect between the exposure and the outcome, thereby improving the reliability of causal inference. Because of the random segregation of alleles at the meiotic stage and the stochastic nature of germline genetic variation at fertilization, MR analyses can avoid confounding factors and reverse causation. In this study, we utilized a two-sample bidirectional MR approach to explore the associations between MDD, SCZ, BD, and thyroid cancer based on statistically pooled data from a genome-wide association study (GWAS). This study aimed to gain insights into the potential link between psychiatric disorders and thyroid cancer and to provide new perspectives and insights for the prevention and treatment of thyroid cancer.
This study is an analysis of previously collected and published public data, including statistical aggregations related to MDD, SCZ, BD, and thyroid cancer, from large public GWASs. Due to the source and nature of the data, no additional ethical review or informed consent was required for this study. Two-sample bidirectional MR analyses were used to assess the causal relationship between psychiatric disorders (MDD, SCZ, BD) and thyroid cancer. We chose psychiatric disorders (MDD, SCZ, BD) as the exposure factor and thyroid cancer as the outcome indicator. Moreover, we conducted a reverse two-sample MR analysis with thyroid cancer as an exposure factor and psychiatric disorders (MDD, SCZ, BD) as an outcome indicator. A flow chart of the MR research design constructed according to this paper is shown in Fig. 1 .

Flowchart of the design of a Mendelian randomized study of the causal association between psychiatric disorders and thyroid cancer. Blue solid lines represent associations between instrumental variables (SNPs) and exposure and between exposure and outcome. The red solid line represents reverse causality. psychiatric disorders include major depression, schizophrenia, and bipolar disorder
Data sources for patients with major depression, schizophrenia, bipolar disorder, or thyroid cancer
The summary-level dataset used for GWASs for MDD in this study was obtained from a meta-analysis of GWAS data conducted by Howard et al. [ 26 ]. It comprises three large-scale GWASs, the Psychiatric Genomics Consortium (PGC), the UK Biobank, and 23andme. Of the three GWASs, only the UK Biobank and the PGC publish summary statistics on genetic variation. The dataset included 500,199 European subjects, including 170,756 cases and 329,443 controls. In the UK Biobank, Howard et al. used a broad definition of depression and asked participants if they reported neurological, anxiety, tension, or depression symptoms to their general practitioner or psychiatrist. At the PGC, Wray et al. diagnosed depression in participants according to international consensus diagnostic criteria (DSM-IV, ICD-9, or ICD-10). See Table 1 for details.
Statistical summary data for SCZ and BD were obtained from the most recent PGC’s GWAS summary statistics. The data for SCZ [ 27 ] are based on a major meta-analysis of multiple groups, including Europeans, East Asians, African Americans, and Latinos, including 76,755 cases and 243,649 control participants. BD [ 28 ] was based on a summary analysis of European ancestry and included 20,352 cases and 31,358 control participants. See Table 1 for detailed information.
To perform two-sample bidirectional analyses, we used independent genome-wide significant single nucleotide polymorphisms (SNPs) as exposure indicators for MDD (50 SNPs), SCZ (217 SNPs), and BD (16 SNPs). The F-statistics of the above SNPs are all greater than 10, indicating that they are strongly correlated instrumental variables (IVs). The detailed information is specified in the Supplementary Material: Tables 1 , 2 and 3 .
Data on genetic variants associated with thyroid cancer were obtained by Deutsches the Krebsforschungszentrum (DKFZ) through GWAS [ 29 ] and included 1080 European participants, including 649 in the case group and 431 in the control group, as detailed in Table 1 . For the bivariate analyses, we used independent genome-wide significant SNPs as indicators of exposure to thyroid cancer (347 SNPs). All 347 SNPs had F-statistics greater than 10, indicating that they were strongly correlated IVs. Specific SNP information is provided in the Supplementary Material: Table 4 .
Selection of genetic instrumental variables
This study was conducted in strict accordance with the quality control steps. First, we selected exposure-related GWAS data and screened SNP loci with genome-wide significance ( p < 5 × 10 − 8 ) for pooled aggregation. Second, to avoid linkage disequilibrium (LD) from affecting the results, we performed a clustering process by setting the parameter (r 2 ) threshold (r 2 < 0.001 and region width = 10,000 kb) to assess LD among SNPs to ensure independence. SNPs need to fulfil three basic assumptions to serve as IVs for exposure factors, and the fulfilment of these assumptions will enhance the testing power and estimation accuracy of IVs: (1) the association assumption: genetic variants are associated with exposure; (2) the independence assumption: genetic variants are independent of confounders between exposure and outcome; and (3) the exclusivity assumption: genetic variants affect the outcome only through exposure [ 30 ]. Next, we extracted summary statistics of eligible SNPs from the outcome GWAS; finally, we determined that the SNPs included in the dataset met the instrumental variable requirements. The palindromic sequences were excluded to ensure that the effects of SNPs on exposure and outcome were from the same allele. This series of steps finalized the identification of SNPs that served as genetic IVs for this study.
Statistical analysis
After coordinating the GWAS effect alleles for MDD, SCZ, BD, and thyroid cancer, we selected three MR approaches. The inverse variance weighted (IVW) test, MR–Egger regression, and weighted median estimator (WME) were used to assess the causal relationship between psychiatric disorders and thyroid cancer risk. The main method of analysis was IVW, while WME and MR–Egger regression were used as complementary methods to IVW estimation, as they provide more reliable estimates under more relaxed conditions [ 31 ]. The Cochran’s Q test was used to estimate the heterogeneity of the causal effects of individual gene variants. If horizontal pleiotropy or heterogeneity is detected, fixed-effects IVW analysis should be chosen, and vice versa for random-effects IVW analysis [ 32 , 33 ]. The IVW method does not take into account the presence of an intercept term and uses the variance of the outcome as the fitting weight. In contrast, the MR–Egger regression method, which is an MR method for assessing the causal effect of genetic variation on the relationship between exposure and outcome, takes into account the presence of an intercept term [ 34 ]. This method corrects for polytropic bias and detects directed polytropy but is susceptible to instrumental variable assumptions. When the Egger intercept of a linear regression is close to zero, it indicates the absence of directional pleiotropy, thus satisfying the exclusivity assumption. The weighted median method is a method that combines data from multiple genetic variants into a single causal estimate and requires that more than 50% of the weights come from valid IVs to obtain a reliable estimate of the causal effect [ 31 ]. To ensure the reliability of the MR estimates, we also detected outliers that may affect our MR estimates by looking at forest plots, funnel plots, scatter plots, and leave-one-out methods.
To test the first hypothesis of correlation, we also assessed the strength of the relationship between IVs and phenotype using the F-statistic (F = beta 2 / se 2 , with beta being the allele effect value and SD being the standard deviation), with F > 10 indicating the presence of strongly correlated IVs [ 35 ].
All of the above MR-related statistical analyses were implemented using TwoSampleMR in R 4.1.1 software.
Three sets of genetic instruments were constructed for the forwards MR study after a series of quality control steps. First, we merged the exposure (MDD)- and outcome (thyroid cancer)-related datasets, and after removing 2 palindromic sequences (rs4936276 and rs4730387), we ultimately included 26 SNPs for analysis. The second set of genetic tools was constructed after the same quality control steps, combining the exposure (SCZ) and outcome (thyroid cancer) datasets and deleting six palindromic sequences (rs12363019, rs217310, rs2470951, rs2944821, rs7709645, rs9925915) before finally including 111 SNPs that were analysed. A third set of genetic instruments was constructed following the same quality control steps, combining exposure (BD) and outcome (thyroid cancer), and after deleting 2 palindromes sequences (rs10455979, rs5758065), and finally included 9 SNPs for analysis. The F-statistics of the above SNPs were greater than 10, indicating that they were strongly correlated with each other (Supplementary Material: Tables 1 , 2 and 3 ).
Three sets of genetic instruments were constructed in the reverse MR study after a series of quality control steps. First, we combined exposure (thyroid cancer) and outcome (MDD)-related datasets, resulting in the inclusion of 331 SNPs for analysis. The second set of genetic instruments was constructed following the same quality control steps, combining the exposure (thyroid cancer) and outcome (SCZ) datasets, resulting in the inclusion of 338 SNPs for analysis. A third set of genetic instruments was constructed following the same quality control steps, combining the exposure (thyroid cancer) and outcome (BD) data and ultimately including 338 SNPs for analysis. The F values of the above IVs were all > 10, indicating reliable results without weak bias.
Mendelian randomization analysis
In our study, we explored the causal relationship between psychiatric disorders (MDD, SCZ, and BD) and thyroid cancer using psychiatric disorders as exposure factors. The results of the IVW analysis showed a significant association between MDD and the risk of thyroid cancer (OR = 3.956 95% CI = 1.177–13.299; P = 0.026), confirming the possibility of an increased risk of thyroid cancer due to MDD. These findings were reinforced by the results obtained by the WME method (OR = 5.563 95% CI = 0.998–31.008; P = 0.050), which were consistent with those of the IVW method. However, the results of the MR–Egger regression (OR = 76.975 95% CI = 0.008-766576.333; P = 0.364) showed that the difference in the effect of MDD and thyroid cancer was not statistically significant (Table 2 ), which may be due to the high false-positive rate of false-negative results from this method. Nevertheless, the IVW and WME methods suggest that MDD may increase the risk of thyroid cancer.
In addition, we found that genetic susceptibility to SCZ was correlated with thyroid cancer (OR = 1.532 95% CI = 1.123–2.088; P = 0.007). The results of the WME method analysis based on the median estimate (OR = 1.599 95% CI = 1.014–2.521; P = 0.043) also support that SCZ may increase the risk of thyroid cancer (Table 2 ).
However, no causal relationship between BD and thyroid cancer was found in any of the MR analyses. In addition, we performed reverse MR analysis, which showed no evidence of a causal relationship between genetic susceptibility to thyroid cancer and psychiatric disorders (MDD, SCZ, and BD), ruling out the possibility of reverse causation (Supplementary Material: Table 5 ).
Sensitivity analysis
For sensitivity analysis, we first tested for heterogeneity of results using Cochran’s Q for IVW and MR–Egger regression. The results showed that the p values of the analyses were greater than 0.05, which indicated that there was no significant heterogeneity in our study. Similarly, the MR–Egger intercept method results also showed no horizontal pleiotropy (all p values greater than 0.05). We also constructed funnel plots and leave-one-out plots. The funnel plot was roughly symmetrical, indicating a relatively low risk of bias and high reliability of the results. A leave-one-out plot was generated to reject SNPs one by one, and the analysis showed that the causal relationship between psychiatric disorders and thyroid cancer was largely not driven by a single SNP. We also examined scatter plots, in which each point represents an instrumental variable. Each horizontal solid line in the forest plot reflects a single SNP estimated using the Wald ratio method. Leave-one-out, scatter, funnel, and forest plots can be found in the supplementary materials.
In the inverse sensitivity analyses, Cochran’s Q test revealed heterogeneity between the effects of thyroid cancer on MDD, SCZ, and BD. Therefore, IVW analysis under a random effects model was chosen to balance the heterogeneity of the results. However, it is noteworthy that no heterogeneity was found in thyroid cancer patients with MDD or SCZ. p values for the MR–Egger intercept method were all greater than 0.05, suggesting that there was no horizontal pleiotropy in the results. Leave-one-out, scatter, funnel, and forest plots can be found in the supplementary materials.
With the increasing prevalence of psychiatric disorders and thyroid cancer, there is an increasing overlap between them, prompting us to delve deeper into their relationship. This study is the first two-sample bidirectional MR study of psychiatric disorders (MDD, SCZ, BD) and thyroid cancer. Our MR study showed a significant causal association between MDD and SCZ and thyroid cancer, whereas no such association was found between BD and thyroid cancer. Reverse MR analysis ruled out the possibility of reverse causation.
MR studies have the advantage of effectively avoiding confounding bias. Because SNPs are randomly assigned at conception, MR is also able to exclude reverse causality effects relative to observational studies, thus enhancing the credibility of causal inferences. We suggest the following possible mechanisms for the positive causal relationship between MDD and thyroid cancer: First, MDD may lead to abnormal functioning of the hypothalamic–pituitary–thyroid (HPT) axis, which in turn affects thyroid hormone levels and thyroid-stimulating hormone (TSH) secretion. TSH is a key factor in promoting thyroid cell proliferation, and abnormal TSH levels may increase the risk of thyroid nodules and cancer. Patients with early-stage MDD may suffer from thyroid and metabolic dysfunction [ 36 ]. Data from the study showed that 26.2% of depressed patients had abnormal thyroid function, 18.3% of whom had MDD, and 62.4% of the study population was female [ 37 ]. The results of a multicentre study by the European Antidepressant Study Group showed that the prevalence of hypothyroidism and hyperthyroidism in patients with MDD was 13.2% and 1.6%, respectively [ 38 ]. These results imply that MDD may regulate thyroid hormone levels through the HPT axis, thereby affecting thyroid function and structure. Abnormal HPT axis function has been the focus of research on neuroendocrine mechanisms in patients with psychiatric disorders, and our findings provide insight into the relationship between genetic susceptibility to MDD and thyroid cancer. Second, MDD leads to elevated peripheral inflammatory marker levels [ 39 , 40 ], which induce chronic inflammation and gene mutations in the thyroid gland. These peripheral inflammatory markers include interleukins (ILs), tumour necrosis factor (TNF), and C-reactive protein (CRP), which can affect thyroid tissues through blood circulation or neuroendocrine pathways. Chronic inflammation can mediate tumour development, and the two are interconnected through endogenous and exogenous pathways. Chronic inflammation of the thyroid gland may contribute to genetic defects through the secretion of high levels of mutagenic agents (e.g., reactive oxygen species and nitric oxide) [ 41 ]. Finally, MDD may be associated with type C personality, which is characterized by abnormal emotional expression and abnormal emotion regulation that may affect the immune system and endocrine function [ 42 , 43 , 44 ]. Some scholars [ 43 ] regard negative emotions as an independent risk factor for the occurrence of thyroid cancer and believe that the persistence or recurrence of depression and anxiety is a stress factor for the human body and that stress causes changes in the cerebral cortex and hypothalamus, which can directly or indirectly suppress the immune system and interfere with the endocrine function of the body [ 44 ], thus affecting the normal synthesis and release of thyroid hormones and triggering thyroid nodules and increasing the likelihood of thyroid cancer.
There may be different aetiologies regarding the genetic susceptibility of patients with SCZ to an increased risk of thyroid cancer. Beginning in the late 19th century, when hypothyroidism was connected with psychiatric disorders, an increasing number of clinical studies have shown a strong independent association between SCZ and hypothyroidism [ 45 ]. A recent community-based cross-sectional study comparing patients with SCZ ( n = 1252) and healthy controls matched for age, sex, socioeconomic status and ancestry ( n = 3756) revealed that the incidence of hypothyroidism in patients with SCZ increased after treatment but not before diagnosis [ 22 ]. Similarly, in observational studies, patients with SCZ are more likely to have abnormal thyroid function after initiating treatment with antipsychotics [ 46 , 47 ]. Thus, the use of antipsychotics may lead to abnormalities in thyroid function, although it is not clear whether the HPT axis can be directly affected. A systematic review and meta-analysis summarizing 19 studies suggested that TSH levels may be reduced at the onset of psychosis and elevated in patients with multiple episodes of psychosis [ 48 ]. Studies have shown that dopamine or dopamine agonists inhibit TSH secretion, and a possible explanation for the elevated TSH levels lies in the fact that antidopaminergic drugs used to treat SCZ inhibit dopamine neurotransmission, which may cause elevated TSH levels [ 49 ]. Thus, there may be a causal relationship between hypothyroidism or elevated TSH levels and the manifestations of SCZ. Thyroid hormones not only play a role in the dopaminergic system but also in the regulation of serotonergic, glutamatergic, and GABAergic networks [ 50 ]. During neurodevelopment, thyroid hormones play a critical role, and their deficiency may severely impair the development of neural tissues, leading to abnormalities and damage in the cerebellar cortex and cerebral cortex [ 51 ]. In the adult brain, thyroid hormone interacts with glial cells to regulate immune responses and neurotransmitter release and to control neuronal metabolism.
However, our study did not find conclusive evidence to support a causal role between genetic susceptibility to BD and thyroid cancer risk. To date, the association between affective disorders and thyroid cancer has not been widely reported. Although previous epidemiologic studies using case–control methods have suggested an association between BD and abnormal thyroid function [ 52 , 53 , 54 ], this topic has yet to be thoroughly investigated. One large meta-analysis reported that thyroid hormones may affect neurodevelopment by modulating the brain’s serotonin system [ 43 ]. The current preferred mood stabilizer for maintenance treatment of BD is lithium, although lithium alters thyroid functional status [ 55 ]. However, little is known about the pathophysiologic role of thyroid hormones in BD, and genetically, our study did not find a direct relationship between BD and thyroid cancer. However, further validation with larger datasets is needed in the future.
Our study has important implications for understanding the potential link between psychiatric disorders and thyroid cancer, as well as providing new ideas and strategies for the prevention and treatment of these common psychiatric disorders. For example, we can reduce the risk of thyroid cancer by screening and treating psychiatric disorders or improve the clinical management of psychiatric disorders by monitoring and regulating thyroid hormone levels. Specifically, we could conduct thyroid function testing and interventions in patients with psychiatric disorders, along with psychological assessment and treatment in patients with thyroid cancer. This integrated approach is expected to mitigate, to some extent, the adverse effects of psychiatric disorders and thyroid cancer on patients’ quality of life and socioeconomic status. In addition, our study provides clues and research directions for in-depth exploration of the potential relationships between MDD and thyroid cancer and between SCZ and thyroid cancer. More experimental and clinical studies are needed in the future to validate our findings and reveal the molecular cellular mechanisms underlying the causal relationship between psychiatric disorders and thyroid cancer.
In conclusion, our study is the first two-sample bidirectional MR study on the causal relationship between psychiatric disorders and thyroid cancer. Although our study provides useful insights for obtaining a deeper understanding of the relationship between psychiatric disorders and thyroid cancer, there are several limitations to consider. First, we used European population-based GWAS data to select IVs and obtain exposure data, which may limit the generalizability and applicability of our results. Second, our IVs were based on the use of a single nucleotide polymorphism-based design, which may not fully capture genetic variability in exposure or outcome. Finally, due to the limitations of the dataset, the number of thyroid cancer patients in the study was relatively small, which may have led to bias.
In summary, our study provides some suggestive evidence that MDD and SCZ are positively associated with thyroid cancer. This finding may have implications for health care policies regarding psychiatric disorders and thyroid cancer. Considering the high prevalence of psychiatric disorders and thyroid cancer in the general population, revealing the causal relationship between psychiatric disorders and thyroid cancer is important for public health policies for early prevention and timely prevention.
Data availability
Major depression:https://gwas.mrcieu.ac.uk/datasets/ieu-b-102/;Schizophrenia;https://gwas.mrcieu.ac.uk/datasets/ieu-b-5099/; Bipolar disorder:https://gwas.mrcieu.ac.uk/datasets/ieu-b-41/; Thyroid caencer:https://gwas.mrcieu.ac.uk/datasets/ieu-a-1082/;
Abbreviations
- Major depressive disease
schizophrenia
bipolar disorder
- Mendelian randomization
instrumental variables
inverse variance weighted
ultrasound-guided Fine Needle Aspiration
genome-wide association study
Psychiatric Genomics Consortium
single nucleotide polymorphisms
Deutsches the Krebsforschungszentrum
linkage disequilibrium
Weighted Median Estimator
hypothalamic–pituitary–thyroid
thyroid-stimulating hormone
interleukins
tumour necrosis factor
C-reactive protein
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72:7–33.
Article PubMed Google Scholar
Haupt S, Caramia F, Klein SL, Rubin JB, Haupt Y. Sex disparities matter in cancer development and therapy. Nat Rev Cancer. 2021;21:393–407.
Article CAS PubMed PubMed Central Google Scholar
Lim H, Devesa SS, Sosa JA, Check D, Kitahara CM. Trends in thyroid cancer incidence and mortality in the United States, 1974–2013. JAMA. 2017;317:1338–48.
Article PubMed PubMed Central Google Scholar
Brainstorm Consortium, Anttila V, Bulik-Sullivan B, Finucane HK, Walters RK, Bras J, et al. Analysis of shared heritability in common disorders of the brain. Science. 2018;360. https://doi.org/10.1126/science.aap8757 .
Tkachev A, Stekolshchikova E, Vanyushkina A, Zhang H, Morozova A, Zozulya S, et al. Lipid alteration signature in the blood plasma of individuals with schizophrenia, depression, and bipolar disorder. JAMA Psychiatry. 2023;80:250–9.
Leber SL, Llenos IC, Miller CL, Dulay JR, Haybaeck J, Weis S. Homer1a protein expression in schizophrenia, bipolar disorder, and major depression. J Neural Transm (Vienna). 2017;124:1261–73.
Article CAS PubMed Google Scholar
Avenevoli S, Swendsen J, He JP, Burstein M, Merikangas KR. Major depression in the national comorbidity survey-adolescent supplement: prevalence, correlates, and treatment. J Am Acad Child Adolesc Psychiatry. 2015;54:37–e442.
Delitala AP, Terracciano A, Fiorillo E, Orru V, Schlessinger D, Cucca F. Depressive symptoms, thyroid hormone and autoimmunity in a population-based cohort from Sardinia. J Affect Disord. 2016;191:82–7.
Gold PW. The organization of the stress system and its dysregulation in depressive illness. Mol Psychiatry. 2015;20:32–47.
Gold PW. Endocrine factors in key structural and intracellular changes in depression. Trends Endocrinol Metab. 2021;32:212–23.
Montero-Pedrazuela A, Venero C, Lavado-Autric R, Fernandez-Lamo I, Garcia-Verdugo JM, Bernal J, et al. Modulation of adult hippocampal neurogenesis by thyroid hormones: implications in depressive-like behavior. Mol Psychiatry. 2006;11:361–71.
Chaker L, Bianco AC, Jonklaas J, Peeters RP, Hypothyroidism. Lancet. 2017;390:1550–62.
Shoib S, Ahmad J, Wani MA, Ullah I, Tarfarosh SFA, Masoodi SR, et al. Depression and anxiety among hyperthyroid female patients and impact of treatment. Middle East Curr Psychiatry. 2021;28. https://doi.org/10.1186/s43045-021-00107-7 .
Duenas OHR, Hofman A, Luik AI, Medici M, Peeters RP, Chaker L. The cross-sectional and longitudinal association between thyroid function and depression: a population-based study. J Clin Endocrinol Metab. 2023. https://doi.org/10.1210/clinem/dgad620 .
Article Google Scholar
Chaker L, Razvi S, Bensenor IM, Azizi F, Pearce EN, Peeters RP, Hypothyroidism. Nat Rev Dis Primers. 2022;8:30.
Zhu GL, Xu C, Yang KB, Tang SQ, Tang LL, Chen L, et al. Causal relationship between genetically predicted depression and cancer risk: a two-sample bi-directional mendelian randomization. BMC Cancer. 2022;22:353.
Park B, Youn S, Yi KK, Lee SY, Lee JS, Chung S. The prevalence of depression among patients with the top ten most common cancers in South Korea. Psychiatry Investig. 2017;14:618–25.
Hartung TJ, Brahler E, Faller H, Harter M, Hinz A, Johansen C, et al. The risk of being depressed is significantly higher in cancer patients than in the general population: prevalence and severity of depressive symptoms across major cancer types. Eur J Cancer. 2017;72:46–53.
Jauhar S, Johnstone M, McKenna PJ, Schizophrenia. Lancet. 2022;399:473–86.
Hakulinen C, Elovainio M, Arffman M, Lumme S, Pirkola S, Keskimaki I, et al. Mental disorders and long-term labour market outcomes: nationwide cohort study of 2 055 720 individuals. Acta Psychiatr Scand. 2019;140:371–81.
Tanskanen A, Tiihonen J, Taipale H. Mortality in schizophrenia: 30-year nationwide follow-up study. Acta Psychiatr Scand. 2018;138:492–9.
Melamed SB, Farfel A, Gur S, Krivoy A, Weizman S, Matalon A, et al. Thyroid function assessment before and after diagnosis of schizophrenia: a community-based study. Psychiatry Res. 2020;293:113356.
Sharif K, Tiosano S, Watad A, Comaneshter D, Cohen AD, Shoenfeld Y, et al. The link between schizophrenia and hypothyroidism: a population-based study. Immunol Res. 2018;66:663–7.
Jurado-Flores M, Warda F, Mooradian A. Pathophysiology and clinical features of neuropsychiatric manifestations of thyroid disease. J Endocr Soc. 2022;6:bvab194.
Smith GD, Ebrahim S. Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32:1–22.
Howard DM, Adams MJ, Clarke TK, Hafferty JD, Gibson J, Shirali M, et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci. 2019;22:343–52.
Trubetskoy V, Pardinas AF, Qi T, Panagiotaropoulou G, Awasthi S, Bigdeli TB, et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature. 2022;604:502–8.
Stahl EA, Breen G, Forstner AJ, McQuillin A, Ripke S, Trubetskoy V, et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat Genet. 2019;51:793–803.
Kohler A, Chen B, Gemignani F, Elisei R, Romei C, Figlioli G, et al. Genome-wide association study on differentiated thyroid cancer. J Clin Endocrinol Metab. 2013;98:E1674–81.
Lawlor DA. Commentary: two-sample mendelian randomization: opportunities and challenges. Int J Epidemiol. 2016;45:908–15.
Bowden J, Smith GD, Haycock PC, Burgess S. Consistent estimation in mendelian randomization with some invalid instruments using a weighted median estimator. Genet Epidemiol. 2016;40:304–14.
Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37:658–65.
Bowden J, Del Greco MF, Minelli C, Zhao Q, Lawlor DA, Sheehan NA, et al. Improving the accuracy of two-sample summary-data mendelian randomization: moving beyond the NOME assumption. Int J Epidemiol. 2019;48:728–42.
Bowden J, Smith GD, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int J Epidemiol. 2015;44:512–25.
Pierce BL, Ahsan H, Vanderweele TJ. Power and instrument strength requirements for mendelian randomization studies using multiple genetic variants. Int J Epidemiol. 2011;40:740–52.
Peng P, Wang Q, Lang XE, Liu T, Zhang XY. Association between thyroid dysfunction, metabolic disturbances, and clinical symptoms in first-episode, untreated Chinese patients with major depressive disorder: undirected and bayesian network analyses. Front Endocrinol (Lausanne). 2023;14:1138233.
Kafle B, Khadka B, Tiwari ML. Prevalence of thyroid dysfunction among depression patients in a tertiary care centre. JNMA J Nepal Med Assoc. 2020;58:654–8.
PubMed PubMed Central Google Scholar
Fugger G, Dold M, Bartova L, Kautzky A, Souery D, Mendlewicz J, et al. Comorbid thyroid disease in patients with major depressive disorder - results from the European Group for the study of resistant depression (GSRD). Eur Neuropsychopharmacol. 2018;28:752–60.
D’Acunto G, Nageye F, Zhang J, Masi G, Cortese S. Inflammatory cytokines in children and adolescents with depressive disorders: a systematic review and meta-analysis. J Child Adolesc Psychopharmacol. 2019;29:362–9.
Panjwani AA, Aguiar S, Gascon B, Brooks DG, Li M. Biomarker opportunities in the treatment of cancer-related depression. Trends Mol Med. 2022;28:1050–69.
Shengshan L, Junyuan L, Wulin Z, Xiaoming C. Advances in the study of chronic inflammation and thyroid cancer. Oncol Prog. 2022;20. https://doi.org/10.11877/j.issn.1672-1535.2022.20.05.03 .
Li-Na G, Yan-Jin L, Jing W et al. Research progress of the correlation between C-type personality and malignant tumor. Mod Prev Med. 2019;46.
Fang C, Kai W, Mingxing X et al. Meta-analysis of risk factors of thyroid cancer base on case -control study. Chin J Endemiol. 2017;36.
Mohammadpour H, Bucsek MJ, Hylander BL, Repasky EA. Depression stresses the immune response and promotes prostate cancer growth. Clin Cancer Res. 2019;25:2363–5.
Feldman AZ, Shrestha RT, Hennessey JV. Neuropsychiatric manifestations of thyroid disease. Endocrinol Metab Clin North Am. 2013;42:453–76.
Vedal TSJ, Steen NE, Birkeland KI, Dieset I, Reponen EJ, Laskemoen JF, et al. Free thyroxine and thyroid-stimulating hormone in severe mental disorders: a naturalistic study with focus on antipsychotic medication. J Psychiatr Res. 2018;106:74–81.
Zhao Y, Wen SW, Li M, Sun Z, Yuan X, Retnakaran R, et al. Dose-response association of acute-phase quetiapine treatment with risk of new-onset hypothyroidism in schizophrenia patients. Br J Clin Pharmacol. 2021;87:4823–30.
Misiak B, Stanczykiewicz B, Wisniewski M, Bartoli F, Carra G, Cavaleri D, et al. Thyroid hormones in persons with schizophrenia: a systematic review and meta-analysis. Prog Neuropsychopharmacol Biol Psychiatry. 2021;111:110402.
Haugen BR. Drugs that suppress TSH or cause central hypothyroidism. Best Pract Res Clin Endocrinol Metab. 2009;23:793–800.
Santos NC, Costa P, Ruano D, Macedo A, Soares MJ, Valente J, et al. Revisiting thyroid hormones in schizophrenia. J Thyroid Res. 2012;2012:569147.
Dezonne RS, Lima FR, Trentin AG, Gomes FC. Thyroid hormone and astroglia: endocrine control of the neural environment. J Neuroendocrinol. 2015;27:435–45.
Hu LY, Shen CC, Hu YW, Chen MH, Tsai CF, Chiang HL, et al. Hyperthyroidism and risk for bipolar disorders: a nationwide population-based study. PLoS ONE. 2013;8:e73057.
Bauer M, Berman S, Stamm T, Plotkin M, Adli M, Pilhatsch M, et al. Levothyroxine effects on depressive symptoms and limbic glucose metabolism in bipolar disorder: a randomized, placebo-controlled positron emission tomography study. Mol Psychiatry. 2016;21:229–36.
Walshaw PD, Gyulai L, Bauer M, Bauer MS, Calimlim B, Sugar CA, et al. Adjunctive thyroid hormone treatment in rapid cycling bipolar disorder: a double-blind placebo-controlled trial of levothyroxine (L-T4) and triiodothyronine (T3). Bipolar Disord. 2018;20:594–603.
Ferensztajn-Rochowiak E, Chlopocka-Wozniak M, Rybakowski JK. Ultra-long-term lithium therapy: all-important matters and a case of successful 50-year lithium treatment. Braz J Psychiatry. 2021;43:407–13.
Download references
Acknowledgements
This analysis benefited from the valuable data sets provided by various researchers and the summary statistics of multiple GWAS shared by the research community. We thank them for their contributions.
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author information
Authors and affiliations.
The School of Clinical Medicine, Fujian Medical University, Fuzhou, 350122, China
Rongliang Qiu & Jinbo Fu
Department of General Surgery, Zhongshan Hospital of Xiamen University, Xiamen, 361004, China
School of Nursing, Fujian University of Traditional Chinese Medicine, Fuzhou, 350122, China
Huihui Lin, Hongzhan Jiang & Jiali Shen
School of Medicine, Xiamen University, Xiamen, 361004, China
You can also search for this author in PubMed Google Scholar
Contributions
Conception and design: QRL, FJB. Development of methodology: JHZ, LHH. Analysis and interpretation of data: SJL, HJX. Writing of the manuscript: QRL, LHH. Study supervision: FJB.
Corresponding author
Correspondence to Jinbo Fu .
Ethics declarations
Ethics approval and consent to participate.
Not applicable.
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Publisher’s note.
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material 1
Rights and permissions.
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/ . The Creative Commons Public Domain Dedication waiver ( http://creativecommons.org/publicdomain/zero/1.0/ ) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
Reprints and permissions
About this article
Cite this article.
Qiu, R., Lin, H., Jiang, H. et al. Association of major depression, schizophrenia and bipolar disorder with thyroid cancer: a bidirectional two-sample mendelian randomized study. BMC Psychiatry 24 , 261 (2024). https://doi.org/10.1186/s12888-024-05682-7
Download citation
Received : 30 November 2023
Accepted : 14 March 2024
Published : 09 April 2024
DOI : https://doi.org/10.1186/s12888-024-05682-7
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
- Schizophrenia
- Bipolar disorder
- Thyroid cancer
BMC Psychiatry
ISSN: 1471-244X
- Submission enquiries: [email protected]
- General enquiries: [email protected]

An official website of the United States government
The .gov means it’s official. Federal government websites often end in .gov or .mil. Before sharing sensitive information, make sure you’re on a federal government site.
The site is secure. The https:// ensures that you are connecting to the official website and that any information you provide is encrypted and transmitted securely.
- Publications
- Account settings
Preview improvements coming to the PMC website in October 2024. Learn More or Try it out now .
- Advanced Search
- Journal List
- Front Psychiatry
Genetics Factors in Major Depression Disease
Depressive disorders (DDs) are one of the most widespread forms of psychiatric pathology. According to the World Health Organization, about 350 million people in the world are affected by this condition. Family and twin studies have demonstrated that the contribution of genetic factors to the risk of the onset of DDs is quite large. Various methodological approaches (analysis of candidate genes, genome-wide association analysis, genome-wide sequencing) have been used, and a large number of the associations between genes and different clinical DD variants and DD subphenotypes have been published. However, in most cases, these associations have not been confirmed in replication studies, and only a small number of genes have been proven to be associated with DD development risk. To ascertain the role of genetic factors in DD pathogenesis, further investigations of the relevant conditions are required. Special consideration should be given to the polygenic characteristics noted in whole-genome studies of the heritability of the disorder without a pronounced effect of the major gene. These observations accentuate the relevance of the analysis of gene-interaction roles in DD development and progression. It is important that association studies of the inherited variants of the genome should be supported by analysis of dynamic changes during DD progression. Epigenetic changes that cause modifications of a gene's functional state without changing its coding sequence are of primary interest. However, the opportunities for studying changes in the epigenome, transcriptome, and proteome during DD are limited by the nature of the disease and the need for brain tissue analysis, which is possible only postmortem . Therefore, any association studies between DD pathogenesis and epigenetic factors must be supplemented through the use of different animal models of depression. A threefold approach comprising the combination of gene association studies, assessment of the epigenetic state in DD patients, and analysis of different “omic” changes in animal depression models will make it possible to evaluate the contribution of genetic, epigenetic, and environmental factors to the development of different forms of depression and to help develop ways to decrease the risk of depression and improve the treatment of DD.
Introduction
Depressive disorders (DDs) comprise one of the most widespread forms of psychiatric pathology. According to the World Health Organization, about 350 million people are affected by a DD. The worldwide prevalence of DDs varies from 3% in Japan to 16.9% in the USA; in most countries, this prevalence ranges from 8 to 12% ( 1 ). It is predicted that, by 2020, DDs will be the second-leading cause of disability throughout the world after ischemic heart disease ( 2 ).
DD entails a number of unfavorable consequences with medical and sociological relevance, and affects significantly the quality of life and adaptive ability. Long-term and severe depression mixed with chronic somatic or neurological conditions might lead to attempted suicide.
Despite the great medical and social significance of DDs, there is no clear conceptualization to explain the causes and mechanisms of DD development. Several theories have been suggested to explain the onset of depression and have been confirmed by biochemical, immunological, and physiological studies. Parallel to well-known “monoamine,” “cytokine,” and “stress-induced” (hypothalamus–pituitary–adrenal (HPA) axis and stress theories) depression models, the phenomena of altered brain neural plasticity and neurogenesis and circadian rhythm desynchronosis (the chronobiological model) have been proposed to explain the onset of depression.
Family and twin studies have provided strong evidence for the contribution of genetic factors to the risk of depression. For instance, a meta-analysis of twin research data shows that the heritability rate for depression is 37% (95% CI: 31%−42%), and data from family studies show a two- to threefold increase in the risk of depression in first-degree offspring of patients with depression ( 3 ). Heritability has also been shown to be especially influential in severe forms of depression ( 4 , 5 ). The illness severity depends on whether DDs are inherited maternally or paternally ( 6 , 7 ).
Since 1978, when the first study devoted to identifying possible candidate genes related to DDs was published ( 8 ), many studies have searched for genes involved in the progression of depression worldwide. Based on the available data on the putative neurobiological mechanisms underlying DDs, more than 100 candidate genes have been analyzed to identify the possible associations between their alleles and the risk of depression onset or its symptoms. The studies of DD pathogenesis have yielded conflicting results. Development of DNA microchip technology has made it possible to conduct genome-wide associations studies (GWASs) to look for risk factors of depression onset, independent of the hypotheses to explain depression pathogenesis available at the time. However, GWASs using large sets of samples, including thousands of patients with different forms of DDs and tens of thousands of patients in meta-analyses, have failed to identify any specific loci responsible for predisposition to DDs. These studies have also not unambiguously defined the biological mechanisms underlying the pathogenesis of the pathology of DDs. This failure to identify clearly the genetic associations and underlying mechanisms indicate that depression is a complicated multifactor heterogeneous psychiatric disorder. It is likely that the predisposition to DDs is determined by the coordinated action of many genes and their interaction with each other and with diverse environmental factors. It is also likely that each gene by itself makes a relatively small contribution to the pathogenesis of the disease ( 9 ).
In this review, we discuss the principal theories to explain the development of depression and the genetic evidence in support of these theories. The final section discusses the results of GWASs and the possible contribution of epigenetic factors to the risk of onset of DDs.
Brief characterization of depressive disorder
Depression (lat. depressio —gloominess, oppression) is a psychiatric disorder characterized by a pathologically low mood (hypothymia) and negative esteem about oneself, one's status in the real world, and one's future ( 10 ). In other words, the major depressive disorder is a complex and heterogeneous illness with an etiopathogenesis that is based upon multiple factors that may act at different levels, e.g., psychological, biological, genetic, and social ( 11 ).
Two current reference classifications provide a clinical description of depression: the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) and the International Classification of Diseases, 10th Edition (ICD-10).
According to the DSM-IV, one of the principal forms of depression is major depressive disorder (MDD). For an appropriate diagnosis, five or more of the following 10 DSM-IV symptoms must be exhibited:
- Depressive mood present continuously for a minimum 2-week period prevalent every day and a larger part of the day
- Pronounced elevated emotional psychomotor activity in children and teenagers
- Diminished ability to feel pleasure and rejoice
- Loss or gain of weight against a marked appetite alteration
- Sleep disturbances: insomnia at night and daytime sleepiness
- Objectively registered psychomotor agitation or motor retardation
- Feeling of weakness, loss of energy, marked fatigue even after minimal effort
- Lowered self-esteem and feeling of worthlessness, loss of self-confidence, ungrounded self-accusation to the extent of delirium
- Diminished ability to think or concentrate, mental slowness, lack of resolution
- Thoughts or actions leading to self-injurious or suicidal ideation.
The abovementioned symptoms must be present continuously for at least 2 weeks and cause disturbance of a person's normal vital activities.
In terms of the manifestation of depressive symptoms, the contemporary ICD-10 classification identifies a number of clinical variants of depression, which are classified according to their severity, the presence of psychiatric symptoms, and recurrence of DDs. Episodes of moderate depression (four or more symptoms exhibited; F32.1) and severe depression without psychotic symptoms (commonly with a number of symptoms, usually with lowered self-estimation and suicidal thoughts and attempts; F32.2), and a recurrent (repeated) DD of moderate or severe degree (F33.2–F33.3).
It is important to note that the main classifications of DDs have some differences and that this must be considered when analyzing the experimental data in studies of depression in humans. It is possible that some of the differences in results between studies will be related to the use of different samples of depressed patients who have been diagnosed according to different clinical criteria. Moreover, in our opinion, it is necessary to distinguish exo- and endogenous DD and analyze this type of DD separately. Exogenous, or reactive, depression is usually triggered by some situational stress, as losing a job, the loss of a member of the family, divorce, or relationship difficulties. As opposed to endogenous depression, exogenous is environmentally caused, and associated with anxiety and mood reactivity, and highly sensitive to psychosocial stressors. Endogenous or melancholic depression is a form of DD unrelated to any pronounced exogenous factors (primary severe somatic illness, illness in a close relative, perceived social problems. In exogenous DD can clearly determine the causes of this disease.
Major hypotheses of pathogenesis of depression: from clinical and biochemical data to candidate genes of depression
The monoamine theory.
The monoamine hypothesis of depression, the first theory historically, was proposed by Joseph Schildkraut in the 1960s. This theory was based on the successful use of iproniazid (monoamine oxidase inhibitor) ( 12 – 14 ) and imipramine (reuptake inhibitor of monoamine neuromediators) for depression ( 15 , 16 ). As proposed by this theory, insufficiency of monoamine neuromediators (serotonin, norepinephrine, dopamine) in definite structures of the central nervous system (CNS) may lead to the development of depression. Detailed analysis of the mechanism of action of these preparations and of later designed tricyclic antidepressants and reuptake inhibitors of monoamine neuromediators confirmed the important role of the imbalance and insufficiency of neuromediators in DDs ( 17 – 20 ). According to the monoamine theory, the synthesis, vesicular transport, and receptors of monoamine neuromediators play an important role in the development of DDs. As a result, the first genetic studies focused on identifying and analyzing polymorphisms in genes associated with serotonin, noradrenalin, and dopamine neurotransmission.
Most studies have analyzed SLC6A4 (previously known as SERT ), which encodes the serotonin transporter that is responsible for the reuptake of serotonin (5-HTT) from the synaptic cleft to the presynaptic neuron and thus plays a role in maintenance of the serotonin level in the presynaptic region. Interest in this transporter also arises from the observation that inhibitors of neural serotonin reuptake are used widely in psychiatry for the treatment of depression, anxiety, and other conditions.
The serotonin transporter is encoded by the solute carrier family 6 member 4 gene ( SLC6A4 ) located on chromosome 17q11.1–17q12 ( 21 ). In the promotor region of SLC6A4 , a 5-HTTLPR (5- h ydroxy t ryptamine t ransporter- l inked p olymorphic r egion) polymorphism was shown to be associated with the availability (the long L allele) or absence (the short S allele) of the 44 bp fragment ( 22 ). The L allele bears 16 GC-rich repeated elements of 20–23 bp, whereas the S allele carries 14 similar repeated units that result from the deletion of the region from the 6th to 8th repeated elements ( 23 ). In vitro studies have shown that the S allele is associated with a lower expression level of SLC6A4 mRNA and lower serotonin transporter expression on membranes and, as a consequence, with a lower ability for serotonin reuptake compared with the L allele ( 23 , 24 ). A number of other rare variants of 5-HTTLPR polymorphism, which contained 15, 19, and >20 repeats, were later reported ( 25 ).
In 2006, the single-nucleotide polymorphism (SNP) rs25531 (A → G) near the 5-HTTLPR polymorphism region was revealed by Hu and coauthors. This polymorphism appears to show linkage disequilibrium with 5-HTTLPR, and the G variant is only found in the L allele carriers. The A → G substitution evokes the appearance of the L G allele, the functional analog of the 5-HTTLPR S allele ( 14 , 26 ). This is because the A → G substitution creates a strong AP2–DNA-binding site (TFBS) which, in turn, suppresses the transcription of SLC6A4 in LG allele carriers ( 13 , 14 ). It appears that up to 15% of the individuals included in previous studies as L allele carriers should have been functionally classified as S allele carriers. This error may have distorted the results and created false-positive or false-negative evidence. Moreover, the situation appears to be more complicated and clearly demonstrates possible problems in interpreting the results of association analysis of individual polymorphic markers inside candidate genes.
In 2008, another SNP, rs25532 (C → T) also localized near 5-HTTLPR, was identified in the promotor region of SLC6A4 . This SNP changes the activity of the 5-HTTLPR/rs2553 combination of polymorphisms. For instance, the L AC allele (the combination of the L allele at the 5-HTTLPR polymorphism with the A and C alleles at the rs25531 and rs25532 polymorphisms) is a variant that ensures a high level of SLC6A4 expression ( 27 ). Further studies revealed additional SNPs, with functionally significant changes, such as G56A in exon 2 and 1425V in exon 9. The 1425V mutation is located in the transmembrane area of 5-HTT, which is important for the formation of the secondary structure of this hydrophobic domain.
A polymorphic region comprising three alleles, Stin2.9, Stin2.10, and Stin2.12, was discovered in intron 2 of SLC6A4 . This variable number tandem repeat polymorphism increases the expression in proportion to the number of repeated copies of the 16/17 bp element (12 > 10 > 9), as determined in the embryonic brain and in human JAR cells ( 28 ). Stin2 alleles respond differently to transcription factors YB-1 and CTCF which, in turn, can be regulated by lithium chloride, which is prescribed for the treatment of bipolar disorders ( 28 , 29 ).
The structure of SLC6A4 may be far more complicated. Recent publications have reported that the expression of this gene is modulated by microRNA mir-16 binding sites in the 3′-nontranslating region of the gene ( 30 ). Therefore, polymorphisms localized within or near microRNA binding sites may be able to exert a strong effect on SLC6A4 expression and, consequently, on 5-HTT functions.
It is possible that the abovementioned complexity of SLC5A4 organization may be one reason for the conflicting results obtained by analyses of the association of this gene's polymorphic variants (primarily in the analysis of the L/S polymorphism of the 5-HTTLPR repeat) with the onset of depression. Meta-analyses of these studies allow no final conclusion to be drawn about the role of this polymorphism in the development of depression. For instance, the meta-analysis conducted by Lopez-Leon et al. ( 31 ) disclosed an elevated risk of DDs in S allele carriers, whereas no similar association was found in carriers of other alleles ( 32 – 34 ). The latest meta-analysis of the results of 23 original studies has shown that the S allele raises the risk of DDs; the risk of depression in S allele carriers is increased 1.14-fold (CI: 1.05–1.24). Nevertheless, the high level of heterogeneity of the data included in this meta-analysis should be noted. In all analyzed models, the association p -value does not reach 0.05. This may be due to the inclusion in the meta-analysis of studies of different size samples, including samples comprising fewer than 50 people ( 35 ).
In the context of the monoamine theory of DD development, analysis of a large number of candidate genes has been performed. They are, in particular, receptor genes for dopamine ( DRD3, DRD4 ) and serotonin ( HTR1A, HTR2A, HTR1B, HTR2C ); genes for noradrenalin ( SLC6A2 ) and dopamine ( SLC6A3 ); genes for the enzymes monoamine oxidase A ( MAOA ), tyrosine hydroxylase ( TH ), tryptophan hydroxylase 1 ( TPH1 ), catechol-o-methyl transferase ( COMT ); and the piccolo presynaptic cytomatrix protein ( PCLO ). For each of these genes, polymorphic variants were identified that were associated with point mutations or tandem repeat polymorphisms. These polymorphisms were analyzed in samples from patients of different ethnicity with DD. As for SLC6A4 , different studies have produced conflicting results, and it seems reasonable not to analyze the results of individual studies but to consider only the meta-analyses that have shown the existence of associations between definite variants of the genes and DD development.
One of the first large-scale meta-analyses of genetic case–control research on DDs was conducted in 2008 by Lopez-Leon and coauthors. The final analysis focused on 20 polymorphisms in 18 genes. The pooled odds ratios (ORs) with 95% confidence intervals (CIs) were calculated. Among the genes of the monoaminergic system, statistically reliable associations were found for SLC6A4 and SLC6A3 ( 31 ).
In another meta-analysis, Gatt et al. ( 36 ) attempted to identify genes associated with DD and common genes shared by the five severe psychiatric disorders: MDD, anxiety (including panic disorders), schizophrenia, bipolar disorder, the attention deficit–hyperactivity syndrome ( 36 ). Table Table1 1 displays the data for the genes whose products are involved in monoaminergic neurotransmission.
Associations shown in meta-analyses between DDs and polymorphic variants of the genes linked to the exchange of monoamine neuromediators * .
It appears that polymorphic variants of the genes that are in some way associated with the monoamine theory of DD pathogenesis can influence the risk of developing depression. However, this influence is not large and cannot be currently regarded as unambiguously proven because of conflicting results of both individual studies and meta-analyses.
Stress as a cause of depressive disorders
Chronic stress and stressful life events early in life are strong proximal predictors of the onset of depression. Although the response to stress implies stability or maintenance of homeostasis, long-time activation of the stress system can cause harmful or even fatal consequences by elevating the risk of obesity, heart diseases, depression, and other disorders ( 37 ). The Hypothalamic–pituitary–adrenal axis and its three main components—hypothalamic neurosecretory cells, pituitary gland, and adrenal cortex—are responsible for adaptation to changed environmental conditions and for mobilization of the organism's reserves during exposure to stress of different etiologies. The HPA system operates in the following way (Figure (Figure1). 1 ). In response to a stressor, neurons in the hypothalamic paraventricular nuclei secrete corticotropin-releasing hormone (CRH), which exerts its action on the hypophysis to initiate the release into the blood circulation of adrenocorticotropic hormone (ACTH), which stimulates the release of corticosteroids, particularly cortisol, from the adrenal cortex. The final hormonal product of the HPA axis, cortisol, binds to mineralocorticoid receptors (type 1) and glucocorticoid receptors (type 2) to form hormone–receptor complexes, which are then transported into the cell nucleus where they interact with specific DNA regions, the glucocorticoid-response elements, to activate the expression of hormone-dependent genes ( 38 ).

The “stress-induced” theory of DD onset is based on the assumption that hyperactivity of the HPA system may be an important mechanism underlying the development of depression after exposure to stress. A number of examples of abnormal functioning of the HPA system during depression favor this hypothesis. First, stressful events during one's life are the strongest factors that can initiate depression onset ( 39 , 40 ). Second, depressed patients frequently show elevated cortisol levels (the human endogenous glucocorticoid) in plasma, urine, and cerebrospinal fluid and corticotropin (ACTH) level in plasma ( 41 ). Depressed patients also exhibit increased size of the hypophysis and suprarenal glands ( 42 ) or decreased function of corticosteroid receptors ( 43 ). Excessive activation of the HPA axis is observed in 50% of depressed people, and continuous administration of antidepressants helps to attenuate this activation ( 44 ).
A large cohort of genes is likely to be involved in the normal functioning of the HPA axis, but only some of these genes have been actively investigated in the context of DDs. The genes most likely to be involved in DDs are those encoding the targets of cortisol and other glucocorticoid hormones secreted during stress. Polymorphic variants of these genes were analyzed in case-control studies and in some cases, associations between these variants and development of DD were shown. For instance, associations between DD onset and polymorphic sites of the genes coding for the GR ( NR3C1 ) and mineralocorticoid receptor (MCR; NR3C2 ) have been reported ( 45 – 47 ). Moreover, postmortem studies by Klok et al. ( 46 ) have shown that MCR mRNA expression is reduced in the hippocampus in depressed patients ( 46 ).
Associations with DD were also shown for genes CRHR1 and CRHR2 , which encode CRH receptors ( 48 ). Lui et al. ( 48 ) reported significant associations between depression and the SNV rs242939 in the CRHR1 gene. These authors also showed that the haplotype formed by G–G–T alleles of the rs1876828, rs242939, and rs242941 was most often represented in patients with MDD compared with controls ( 48 ). Szczepankiewicz et al. ( 49 ) found associations between DD and the SNVs rs4076452 and rs16940655 of CRHR1 gene ( 49 ). Xiao et al. ( 50 ) also reported associations between the rs242939 polymorphism of CRHR1 gene and recurrent depression ( 50 ).
We note that some of the associations described earlier were included in the meta-analyses by Lopez-Leon et al. ( 31 ) or Gatt et al. ( 36 ). These authors did not find any reliable associations between DDs and polymorphisms in the genes involved in the functioning and regulation of the HPA axis ( 31 , 36 ).
Disturbance of neurogenesis and neuroplasticity
A large body of experimental data has recently provided evidence of a link between the development of depression with disturbance of normal neurogenesis during brain ontogenetic development and decreased neurogenesis of the adult brain. These effects are thought to be caused by metabolism disturbance of neurotrophic factors, primarily the brain-derived neurotrophic factor (BDNF) in nervous tissue ( 51 – 57 ).
BDNF is abundantly expressed in the adult brain's limbic structures. Some data have shown a connection between the BDNF-mediated signaling pathway and the functioning of serotoninergic neurons. For example, BDNF maintains the survivability and differentiation of serotoninergic neurons, and serotoninergic transmission exerts a strong influence on BDNF expression ( 58 – 60 ).
A functional missense polymorphism rs6265 (GI96A) was described in BDNF that is associated with the substitution of methionine (Met) with valine (Val) in codon 66 (Val66Met). The Met allele was shown to cause disturbed maturation of the protein and to be associated with decreased BDNF activity ( 61 ), which might be caused by two different mechanisms: the infective transport of the mutant protein over the regulatory secretory pathway and the defective transport of BDNF mRNA to dendrites ( 62 ). Val66Met is a frequent polymorphism whose frequency of alleles is determined by ethnicity. Met allele frequency is 25–32% in European populations and reaches 40–50% in Asian populations ( 63 ).
Some studies have reported an association of the Val66Met polymorphism in BDNF with DD onset ( 64 – 66 ), but different authors attribute the risk of DD to the effect of different allelic variants. For instance, Frielingsdorf et al. ( 66 ) showed that Met allele homozygotes are at significantly increased risk of MDD ( 66 ), whereas Ribeiro et al. ( 65 ) defined the Val allele as an allele for the risk for depression ( 65 ). This inconsistency motivated the publication of a meta-analysis that combined the results of 14 original studies, but this analysis did not confirm an association between the polymorphic variant in BDNF with DDs ( 63 , 67 ). The effect of this polymorphism may have been observed only in interaction with other polymorphic systems, such as with 5-HTTLPR polymorphisms or after exposure to severe stress ( 68 , 69 ).
The cytokine theory
The brain was earlier thought to be an “immune-privileged” organ that is protected from circulating immune cells by the blood–brain barrier. It is now well known that immune system cells can infiltrate into nervous tissue. Pro- and anti-inflammatory signals can be transmitted to nervous system from peripheral areas. Also, cytokines and their receptors can be produced in the CNS by astrocytes, microglia ( 70 ) and, in some cases, neurons. These molecules are believed to participate in the processes of neuronal development, plasticity, synaptogenesis, and tissue repair.
The hypothesis of bidirectional communication between the immune system and the CNS was suggested in the 1990s. According to this hypothesis, the immune system can interact with the CNS as well as being involved in neuropathological processes. In 1999, Maes M . proposed the inflammatory response system (IRS) model of depression, which claimed that the occurrence of depression depends on activation of the IRS. According to this model, depression can be regarded as a psychoneuroimmunological disease in which peripheral activation of the immune system through the release of anti-inflammatory cytokines can cause the various behavioral, neuroendocrine, and neurochemical changes observed in this disorder ( 71 ). This theory was later extended and is now referred to as the “cytokine theory.”
Cytokines constitute a heterogeneous class of mediator molecules produced as regulators of the immune response by immunocompetent cells such as lymphocytes and microphages. Cytokines can be classified into two groups: proinflammatory and anti-inflammatory cytokines. Proinflammatory cytokines are either immediately or indirectly involved in the inflammatory process: interleukin 1 (IL-1), IL-2, IL-6, and IL-12; interferon γ (IFNγ); and tumor necrosis factor α (TNFα). Anti-inflammatory cytokines include IL-4, IL-10, and IL-13, which suppress the immune response and thereby prevent both cell activation and the production of proinflammatory molecules. Some cytokines, such as IL-8, exert both the pro- and anti-inflammatory functions, according to their concentration.
Immunological changes during depression and psychiatric side effects caused by the use of cytokines in the treatments for hepatitis and cancer provide evidence in favor of this theory. For instance, stressors increase the expression of proinflammatory cytokines such as IL-1β, TNFα, and IL-6) in blood and brain ( 72 , 73 ). DD patients display increased granulocyte and macrophage counts in peripheral blood ( 74 ).
In addition to associations with the depression, associations between inflammatory markers and some symptoms, such as apathy, cognitive dysfunction ( 75 ), and impaired sleep ( 76 ), have been reported. For instance, sleep impairment in depressed patients is associated with increased levels of IL-6 and the soluble forms of intercellular adhesion type 1 molecules in plasma ( 76 ) and with the activation in blood cells of nuclear factor-κB (NF-κB), the principal transcription factor involved in initiation of the inflammatory response ( 77 ). In other studies, about 50% of the patients chronically administered IFNα developed symptoms of depression, which further supports the role of inflammation in DD pathogenesis ( 78 , 79 ).
We note, however, that the average cytokine levels in the plasma are elevated only slightly in DD patients compared with healthy people and that these levels are sometimes close to the physiological norm. In autoimmune or infectious diseases, the cytokine levels increase considerably more. Therefore, DD is not considered as a typical autoimmune disease ( 74 ).
IL1B is located at locus 2q14.1 on chromosome 2. Several SNPs have been reported for this gene, and two—rs1143627 (−31T/C) and rs16944 (−511C/T)—are associated with DD. Some studies have reported an association between rs16944 (−511C/T) and depression onset and with a positive response to treatment with some antidepressant medications ( 80 – 83 ). Borkowska et al. ( 81 ) reported a positive association with recurrent depression ( P = 0.064) for the polymorphic haplotype comprising the rs1143627 SNP C allele and the rs16944 SNP T allele ( 81 ). By contrast, Yu et al. ( 82 ) did not confirm an association of the rs14944 SNP with major depression, although the severity of depression symptoms was elevated in allele C homozygotes ( 82 ). Similarly, Hwang et al. ( 84 ) failed to find any reliable associations of rs16944 SNP with senile depression or the severity of depression symptoms ( 84 ).
The SNPs rs1143627 (−31T/C) and rs16944 (−511C/T) are found in the promoter region of the gene and can affect the expression of this gene, which influences the IL-1β level. Chen et al. ( 85 ) showed that SNP rs1143627 localizes in the TATA box area in the IL1B promoter and that the T allele of this polymorphism is associated with increased production of IL-1β ( 85 ). However, other investigations have failed to find any reliable associations of SNP rs1143627 with IL-1β production in vitro , although one study found an association between elevated IL-1β expression in vivo and the C allele ( 86 ). Data concerning the influence of rs16944 (−511C/T) on IL1B expression are also conflicting. For instance, Hall et al. ( 87 ) showed that this SNP influences the expression directly ( 87 ). On the other hand, some data support the concept that the −511C/T polymorphism influences the expression of the gene only when acting in concert with the −31T/polymorphism ( 85 ).
Therefore, the current data concerning an association of polymorphic variants of the IL1B promoter area do not allow any unambiguous conclusions to be drawn about the role of this gene in the development of depression.
Another actively studied gene, IL6 , encodes the proinflammatory cytokine IL-6 and is located at the 7p15.3 locus. The functional polymorphism rs1800795 (174 G/ C) is located in the gene's promoter region and influences gene expression both at the mRNA and protein levels. The IL-6 level is lower in C allele carriers under conditions of immune system activation ( 88 ). No association of this polymorphism with major depression, depression in children, or postbrain stroke depression has been found ( 89 – 91 ). However, this polymorphism has been reported to affect the progression of depressive symptoms in hepatitis C patients administered the immunomodulators IFNα and ribavirin ( 92 ). Another interesting observation in that study was the interaction between the 5-HTTLPR polymorphism in the serotonin transport gene ( SLC6A4 ) and rs1800795. A “protective” effect of the 5-HTTLPR polymorphism was observed only in the presence of the low-expressing genotype for IL6 ( CC ). Consequently, in the genetics of depression, the transition from the analysis of individual polymorphic variants to the analysis of their combinations including two, three, or more loci seems to be very important.
The circadian rhythm theory
Circadian rhythms oscillate with ~24-h periodicity and are responsible for regulating a wide variety of physiological and behavioral processes. Endogenous cyclic oscillations are regulated in humans and other mammals by the circadian pacemaker—the suprachiasmatic nucleus (SCN) neurons of the anterior hypothalamus ( 93 ). The circadian pacemaker can change its pattern so that the circadian rhythm may be advanced, delayed, or remain constant in various pathological states or when affected by different pharmacological preparations and hormones; for instance, melatonin regulates the function of the biological clock through melatoninergic receptors residing in the hypothalamic SCN ( 94 ). At the cellular level, the “molecular clock” refers to a network of “clock” genes, which are transcriptional regulators organized in a feedback-sustained transcription–translation network. This mechanism helps maintain the rhythmic expression of target genes during a 24-h cycle ( 95 ). The basis of the molecular clock is the negative feedback loop, in which the expression of PER and CRY proteins is inhibited by their interactions with the transcriptional factors CLOCK and BMAL1 and by blocking their binding to E-box regulatory elements in the promoters of the genes for PER and CRY protein family members. Posttranslational modifications of the molecular clock system components by signal molecules such as casein kinase δ/ε and glycogen synthase kinase 3-beta play important roles in the maintenance of circadian rhythms ( 96 ).
Sleep disorders (early awakening, insomnia, and inability to resume sleep after waking) are observed in 80–90% of patients with depression, and insomnia is regarded as a risk factor for the onset of depression ( 97 ). Therefore, disturbance of the normal functioning of circadian system proteins may play a role in DDs. Additional evidence for the role of circadian rhythm genes in DD onset was provided by studies of two familial syndromes of sleep disturbance: familial advanced sleep-phase syndrome and delayed sleep-phase syndrome (DSPS). Both syndromes are caused by mutations in the genes PER2, CKie, PER3 , and CLOCK , which encode circadian system proteins ( 98 ). People with these mutations frequently experience depressive symptoms. In addition, people with a history of depression have elevated expression of the circadian system genes CLOCK, PER1 , and BMAL1 compared with healthy volunteers ( 99 ). Case–control studies have shown associations between depression onset and polymorphic variants of the genes encoding circadian system proteins, including BMAL1, CLOCK, NPAS2, PER3, CRY1 , and TIMELESS . However, none of these genes was confirmed in the meta-analysis ( 100 ).
Other candidate genes
Investigations of the candidate genes selected according to the principles of DD etiopathogenesis theories have identified only five genes whose polymorphic variants are reliably associated with DD onset according to the results of meta-analyses (Table (Table1). 1 ). However, all of these genes have been discussed only in terms of the monoamine theory of depression.
The association research is not confined solely to the DD-related candidate genes studied in terms of DD etiopathogenesis theories. Other genes have also been actively studied, primarily those analyzed for other neurological and psychiatric disorders. Moreover, these studies have revealed a large amount of reliable associations that have passed through testing in large meta-analyzes (Table (Table2). 2 ). These association studies have revealed genes encoding functionally diverse proteins, from chondroitin sulfate biosynthesis enzymes to the key enzyme of the renin–angiotensin system (RAS). On the one hand, this may reflect the limited understanding of DD etiopathogenesis, although the currently known associations suggest new hypotheses. On the other hand, many of the associations revealed are in some way linked to neurogenesis and neuroplasticity processes, and DDs may be regarded as disorders linked to disturbance of neural tissue ontogenesis.
Association shown by meta-analyses between DD and polymorphic variants of genes not linked with the general hypotheses of DD etiopathogenesis.
According to Gatt and coauthors with modifications ( 36 ) .
Associations between DD and polymorphic variants of the genes angiotensin-converting enzyme ( ACE ), apolipoprotein E ( APOE ), and methylenetetrahydrofolate reductase ( MTHFR ) are of special interest. These genes have been discussed in the context of another theory of DD etiopathogenesis—the vascular theory—which postulates that DD occurs because of disturbance in the blood supply to neural tissue. According to this theory, vascular disorders can cause DD as well as other mental illnesses such as schizophrenia and manic–depressive psychosis. It is possible that there is a continuum of these diseases, which are separated by only a thin line; for example, some clinical subtypes of depression are characterized by marked psychotic symptoms (F32.3 in ICD-10). Presumably, there may be a complex link between genetic variants and the occurrence of various pathologies along the continuum. For example, genetically determined vascular disorders provoke increased risk for different mental disorders. Progression of a particular disease (e.g., schizophrenia, depression, manic–depressive psychosis) may be determined by other genetic factors, such as those associated with neuromediator functions.
Genome-wide association analysis of depressive disorder
As noted above, >20 genes have been associated with DD onset and confirmed by meta-analyses. In most cases, these associations involve genes that are not directly linked to the general theories of depression ethnopathogenesis. Association studies appeared to be connected with transition to genome-wide methods of association analysis without any suggestions about the genetic risk factors of depression.
In the first stage of GWASs, families with members that have experienced multiple depression events, severe course of the disorder, or an early age at its clinical onset were analyzed with special interest in patients with rare monogenic forms of depression. The results of these studies are summarized in Table Table3. 3 . These studies have reported associations with extended genomic regions (even as long as full-length chromosomes), and the identification of the candidate genes seems to be provisional. This identification mainly based on the DD candidate genes mapped earlier in these genome regions.
Mapping the loci associated with predisposition to different forms of depression in family studies using SNP panels of DNA markers.
GWASs have been used increasingly in the past decade to identify loci that control complex traits. In this analysis, as many as hundreds of thousands to several millions of SNPs distributed over the whole genome are identified in groups of persons having a particular trait of interest. Analysis of the genotype–phenotype associations makes it possible to establish a link between the allelic variant in some particular region of the genome with the trait studied. The principal difference between GWASs and candidate gene studies using the case–control method is that there is no preliminary hypothesis to explain the contribution of polymorphic variants of genes to the development of a pathology of interest. However, for a study to achieve statistically significant results, its algorithm requires very large samples of both patients and healthy persons. It can be extremely difficult to achieve clinical homogeneity in very large samples, especially when studying psychiatric diseases because there is always a subjectivity factor affecting the diagnostic accuracy in the relevant international classifications with almost no instrumental methods for assessing the patient's condition.
A number of studies have searched for loci associated with MDD or individual symptoms of depression. The results are summarized in Table Table4. 4 . This table focuses mainly on those studies that analyzed primarily the risk of depression as a disease and not the endophenotypes (e.g., clinical onset age, severity of particular symptoms, patients' responses to therapy). As well, Table Table4 4 includes the most statistically significant results from the analyzed articles.
Genome-wide association studies of major depressive disorders (MDDs) and recurrent depressive disorders (RDDs).
The first GWAS of a large representative sample (1738 DD patients, 1802 controls) was reported by Sullivan et al. ( 106 ). In this study, no association with any of SNPs achieved the value of genome wide significance. The maximum significance was found for the rs2715148 ( p = 7.7 × 10 −7 ). Also, in this genomic region near PCLO gene 10 more SNPs were associated with DD with relatively low significance ( p = 10 −5 -10–6). They were mapped to a 167 kb region where PCLO was located ( 106 ). PCLO protein localizes in the cytoplasmic matrix of the presynaptic active zone and plays a significant role in brain monoaminergic neurotransmission. A possible role of this region in depression onset was confirmed by Hek et al. ( 115 ), who showed an association between the rs2522833 SNP in PCLO and DD in a population-based study from the Netherlands ( 115 ). Aragam et al. ( 113 ) found a close statistically significant association between DD development for the rs2715148 SNP ( P = 5.64 × 10 −7 ) in PCLO in women ( 113 ). This study found another SNP in LGSN that was associated with DD occurrence in men (rs9352774, P = 2.26 × 10 −4 ). This gene is actively expressed in the human crystalline lens and encodes a protein related to GS-I and, to a lesser degree, to GS-II glutamine synthetases. This protein may play a role in glutamate exchange in both the retina and the nervous system.
A role of glutamate in DD was found in a GWAS conducted by Rietschel et al. ( 109 ). They found an association between DD and the rs7713917 SNP ( P = 5.87 × 10 −5 ) located in a putative regulatory region of HOMER1 , which encodes proteins involved in glutaminergic processes via interaction with the metabotropic glutamate receptors mGluR1 and mGluR5.
We reiterate that the associations discovered in most GWASs did not attain a genome-wide significance level, primarily because of the genetic architecture of complex traits predisposing to depression. Adjustments of the genome-wide significance level are very rigorous, and we believe that SNP markers with a probability value close to the genome-wide threshold level should also be considered.
Some studies have achieved a genome-wide significance level. Kohli et al. ( 112 ) were the first to report an association between DDs and the rs1545843 SNP in SLC6A15 (solute carrier family 6, neutral amino acid transporter, member 15) in a recessive model of the effect of this polymorphism on the risk of DDs ( 112 ). This gene encodes the neutral amino acid transporter, and different rs1545843 alleles were shown to have different SLC6A15 expression levels in the hippocampus of epileptic patients. The authors presented additional evidence to support the involvement of this association and showed that the presence of the risk allele correlated with lower SLC6A15 expression in the hippocampus, smaller hippocampus volume, and neuronal integrity in vivo . Lower expression of Slc6a15 was also observed in the hippocampus of mice with elevated chronic stress susceptibility.
Kohli et al. ( 112 ) reported abundant data in support of the association between SLC6A15 and DD. However, subsequent GWAS disclosed no significant associations with this gene. The data obtained in GWASs are often not reproducible, and only one gene, PCLO , appeared to be associated with DD in two GWASs.
The Psychiatric Genomics Consortium (PGC) performed a meta-analysis of GWAS data. Unlike the conventional meta-analyses, which summarize the statistical data for each constituent analysis examined, the PGS study brought together and examined individual genotypic and phenotypic data from patients from different research centers. The PGS published the results of its genome-wide comparative analysis of 9240 samples collected from DD patients and 9519 samples from a control group of nine European populations ( 122 ). However, in the PGS analysis, none of SNPs identified in earlier studies achieved a genome-wide significance level. The SNPs with the most significant values were rs11579964 ( P = 1.0 × 10 −7 ), which mapped near CNIH4, NVL , and WDR26 , and rs7647854 ( P = 6.5 × 10 −7 ), which mapped near C3orf70 and EHHADH . A subsequent replicative study conducted using an independent sample (6783 patients with MDD and 50,695 controls) did not confirm the associations mentioned.
Therefore, no locus has been shown to be consistently associated with a DD at a whole-genome significance level. Associations shown in independent samples have also not been reproduced. This lack of significance and reproducibility may reflect the particular features of the GWAS methodology, which has focused on polymorphic sites with a high minor allele frequency (>5%) in the associative analysis. These frequent polymorphic variants themselves are probably not pathogenically essential, but there may be disequilibrium linkages with rare variants of genes associated with DD pathogenesis. These rare variants may be specific for different populations. As a result, any association between the disease and a frequent polymorphic site may be found in one sample and may reflect the disequilibrium linkage of this polymorphic site with a rare, pathogenically significant variant in that sample. However, the pathogenically significant site may be missing in another sample and, as a consequence, no association of frequent polymorphism with DD occurrence will be found. In addition, the important role of rare genomic variants (a frequency <1%) has been reported in association with other mental disorders, such as schizophrenia and autism ( 123 , 124 ).
To overcome these problems, transition from the analysis of polymorphic DNA markers using microarrays to low-coverage DNA sequencing may provide a new direction for research to identify DD-associated genetic variants. The first study of this kind was conducted within the CONVERGE Project ( 125 ) and included genome sequencing with an average coverage of 1.7 × in >9000 Chinese females; 5000 females out of this group were patients with melancholic depression, which is recognized as a more severe form of depression. This study found two loci bearing an association at a 10 −8 significance level: one on the 5′-side of SIRT1 (SNP rs12415800) and the other in an LHPP intron (SNP rs35936514). This association was confirmed in an independent sample of melancholic Chinese women, and the significance values combined for the two samples were 2.53 × 10 −10 for SIRT1 and 6.45 × 10 −12 for LHPP . It is important to note that both associated SNPs occur frequently (e.g., the minimal allele frequencies were 45.3 and 26.2%), yet neither is included in the microarrays used widely for SNP marker typing and, therefore, may have been ignored in earlier GWASs.
Further analysis of the data in this project showed that frequent SNPs accounted for 20–30% of the DD risk dispersion, which suggested that the heritability of DD is evenly distributed over all chromosomes with preferential localization of DD-associated SNPs in both the coding and the 3′-untranslated areas of genes. DD patients showed an elevated frequency of unique mutations in gene coding regions, primarily in the genes actively expressed in nervous tissue ( 126 ).
Importantly, this study included a specific ethnic group (Han Chinese), which is sufficiently homogeneous, and only females, who show a higher heritability level as mentioned above, with a severe form of DD. This design included a more rigorous approach to inclusion of samples and consideration of factors such as the patients' sex, clinical DD variation, clinical onset age, and other factors that can affect the risk of disease and its progression. However, these factors may exert no influence on the risk of DD development; for example, the clinical onset age was recently shown to not affect the association analysis results in the Chinese CONVERGE sample ( 127 ).
Another study also found that ethnicity was important ( 128 ). That study included a combined analysis of the results obtained in the CONVERGE investigation of Chinese and of studies conducted by the PGC in different European populations. These studies found that some SNPs influence the risk of DD onset in both ethnic groups mentioned but, at the same time, detected a set of SNPs specific to each ethnic group. The highest contribution of genetic factors in both ethnic groups was observed in females and in recurrently depressed patients.
Powers et al. ( 116 ) attempted to include environmental factors into GWASs ( 116 ). They included as a factor stress-provoking events when including case–control pairs of patients in the study—a method referred to as propensity score matching . This analysis allowed them to reduce the heterogeneity of the samples with regard to the stress factor and to compare DD patients and healthy controls exposed to similar stressors.
The genetic structure of depression appears to be extremely complicated and involves a large number of loci, which cause various phenotypic effects and display complex interlocus interactions. Studies of the genetic structure suggest the need for a transition from the analysis of individual SNPs to that of sets of SNPs and, finally, to include a polygenic risk score, as used in genetics research of schizophrenia ( 129 ).
To address similar problem, a strategy for studying gene networks created by uniting signals from numerous SNPs and subsequent functional analysis of the signaling and metabolic pathways have been used with success. This approach provides for an increase in the power of comparative analysis of weak signals from numerous loci. The study by Song et al. ( 130 ) is an example of such an analysis. On the basis of a GWAS of samples from European cohorts, the authors conducted a search and analysis of DD-linked SNPs and genes with these SNPs to discover signal pathways linking these genes to each other ( 130 ). Five resulting signal paths were found to play a role in DD pathogenesis. Three of them were claimed to be connected in some way with the negative regulation of gene expression (GO:0016481, GO:0045934, GO:0010629) and were related to some DD-associated SNPs: rs3213764 in ATF7IP ; rs2301721 in HOXA7 ; rs6720481 in LRRFIP1 ; rs2229742 in NRIP1 .
Okbay et al. ( 118 ) and Hyde et al. ( 131 ) offered an alternative approach for sampling ( 118 , 131 ). To diagnose DDs, they compiled a questionnaire to be completed by the respondents. Depression was diagnosed on the basis of the respondents' answers to the questionnaire with no clinical diagnosis by a psychiatrist. Although the accuracy of the diagnosis may be questioned, the questionnaire included questions on a wide range of phenotypic traits, and respondents could not associate them with any diagnoses. Data from biobanks or mass genotyping services such as 23 and Me allowed them to markedly increase the sample size. For example, the study by Hyde et al. ( 131 ) included >450,000 individuals, and analysis of their questionnaire data allowed them to diagnose depression in about 120,000 participants ( 131 ). Samples of this size are an order of magnitude greater than those included in the PGC studies or CONVERGE Project, and help to minimize the problems caused by DD diagnostic errors. The authors managed to identify 17 SNP markers in 15 loci whose significance level was >5 × 10 −8 , which reflects the size of the sample analyzed. The DNA markers detected differ from those associated with DD in the PGC studies, although they both analyzed samples of European origin. Therefore, the problem of the reproducibility of results obtained in the GWASs remains to be solved.
A possible way to solve this problem is to conduct a meta-analysis of GWA studies. This analysis was carried out by Wray et al. ( 121 ). This meta-analysis identified 44 independent loci that were statistically significant ( P < 5 × 10 −8 ). Of these loci, 30 are new and 14 were significant in a prior study of MDD or depressive symptoms, and 6 shared loci with schizophrenia. Thus, the increase in sample sizes in the meta-analysis, on the one hand, allows the confirmation of the results obtained earlier with GWAS for the previously described loci associated with MDD. On the other hand, it increases the power of the study, by increasing the sample size, making it possible to identify new loci associated with the MDD.
Several methods were proposed for calculating of genetic risk score (GRS): simple count genetic risk score (SC-GRS), odds ratio weighted genetic risk score (OR-GRS), direct logistic regression genetic risk score (DL-GRS), polygenic genetic risk score (PG-GRS) and explained variance weighted genetic risk score (EV-GRS). Currently, the most widely used method is polygenic risk score (PGRS) ( 132 ). This approach has been used to obtain evidence of a genetic effect even when no single markers are significant, to establish a common genetic basis for related disorders, and to construct risk prediction models ( 133 ). Currently, alternative approaches to statistical analysis of GWASs data are proposed, where the analysis is not of individual DNA markers, but their combinations. Recently, several papers have been published using PGRS for the MDD and other psychiatric disorders ( 134 – 136 ). The possibility of using the PGRS to evaluate the cumulative contribution of several polymorphic variants of genes to the formation of endophenotypes of MDD was demonstrated. Whalley et al. ( 135 ), using the PGRS, divided the MDD into two subtypes, one of which is close to schizophrenia ( 135 ).
Conclusions
Summing up the last quarter-century of investigation of the role of genetic factors in DD onset and progression, we note the polygenicity of inherited diseases with no pronounced effect of the principal gene. This has been made clear by recent studies undertaken in the CONVERGENCE and PGC studies and by analysis of candidate genes, which show that each of the genes implicated probably make a small contribution to DD progression. The important role of intergenic interactions has not been studied to date and will require new methods to analyze the data of association studies by including the contribution of combinations of two or more polymorphic DNA markers.
As noted above, DD typically has a high phenotypic heterogeneity, which may be manifested in differing severity of the main symptoms, and this heterogeneity limits the use of association studies. An approach to analyzing very large samples with minimally rigid assessment criteria for the DD phenotype (an extreme example is the variant identified by the 23andMe company associated with self-diagnostics of DD) is one possibility, but this must be supplemented by analysis of small and clinically highly homologous samples. This kind of analysis may not clear up the uncertainty about the genetics of DD on the whole, but it will make it possible to identify certain genetic variants underlying individual subphenotypes of DD.
A small but growing body of evidence suggests that mitochondrial dysfunction may play a role in the development of MDD. MDD was found to be associated with an increased production of mtROS, which could indicate a dysfunction of mitochondria ( 137 , 138 ). Gardner et al. ( 139 ) found a decrease of mitochondrial ATP production rates and mitochondrial enzyme ratios in the muscle of patients with major depressive disorder and chronic physical conditions compared to controls ( 139 ). As well, decreased levels of an important part of electron transport chain, i.e., CoQ10, were found in the serum and peripheral blood mononuclear cells received from patients with MDD, which may also indicate a dysfunction of mitochondria ( 140 ).
Currently, however, there have been few studies on the role of genetic variants in mitochondrial DNA associated with MDD. A deletion in mtDNA in a child was associated with mitochondrial disease symptoms and with mild-moderate unipolar depression ( 141 ). Sequeira et al. ( 142 ) have analyzed post-mortem brain samples from human subjects and were failed to show associations of the mitochondrial haplogroups and major depression. However rare homoplasmic mutations with possible functional consequences were reveled in major depression cases, in the ATP synthase 8 (ATP8), ATP synthase 6 (ATP6), ND5 and cytochrome b (CYTB) genes, while another subject with depression demonstrated subthreshold heteroplasmy rate at a variant in the displacement loop (D-loop) part of mtDNA ( 142 ). Veronese et al. ( 143 ) found no significant associations between specific mitochondrial haplogroups and depressive symptoms either ( 143 ). Thus, changes in the functioning of mitochondria may be caused both by an abnormality of the mitochondrial DNA, and by variants of nuclear genes that encode mitochondrial proteins. The meta-analysis conducted by Huo et al. (141) identified SCL25A37 as a novel MDD risk gene, and Zhang et al. ( 144 ) have showed that a haplotype T-C consisting of rs12457810 and rs12964485 in the 5'-upstream region of NDUFV2 may be a protective factor for the development of MDD in Han Chinese ( 144 , 145 ).
It is also important to supplement the association studies of inherited genome variants with analysis of dynamic modifications occurring during DD. There is much interest in the epigenetic changes that can modify a gene's functional status without changing its coding sequence. These epigenetic modifications can be caused by the action of different factors and can be stably inherited after disappearance of the factor causing the change. These epigenetic factors primarily involve DNA methylation and histone modification (methylation and acetylation). In recent years, several studies have analyzed changes in DNA methylation in DD. The first genome-wide analysis of methylation profiles in DD was by Sabuncian et al. ( 146 ), who assessed DNA methylation in postmortem frontal cortex material from DD patients and healthy people. In a number of regions, methylation differed reliably between healthy individuals and DD patients ( 146 ). A subsequent replicative investigation confirmed this modification of the methylation status in DD patients of the proline rich membrane anchor 1 gene, PRIMA1 , which codes for the protein responsible for the assembly of acetylcholine esterase into tetramers and its “anchorage” in the neuron cellular membranes ( 147 ). This gene has not been mentioned in association with DD onset. Methylome changes were reported in peripheral blood of twins discordant with regard to DD occurrence. This study also reported on associative studies showing that, in some cases, the changes were related to genes linked to the development of the disorder (e.g., ZBTB20, AGTPB1, TBC1D8 , and CLSTN1 ) ( 148 ). A number of studies have focused on associations between DD and histone methylation or acetylation ( 149 , 150 ). Changes in lysine methylation of histone K27H3 were found postmortem in the BDNF promotor region in the prefrontal and frontal cortex of DD patients, and these changes correlated well with the expression level of BDNF . However, in this review, we do not analyze in detail the role of epigenetic factors in the development of the pathogenesis of DD.
Investigation of changes in the epigenome, transcriptome, and proteome in DD is probably limited by the nature of this disease and the need for brain tissue, which is possible only postmortem . Researchers must work with an extremely limited number of samples from patients, many of whom are on long-term treatment for both DD and somatic conditions. Therefore, to understand fully the entire process involved in DD onset and progression, studies of DD pathogenesis must be supplemented with experiments using different animal models of depression, which would permit an evaluation at different levels of the nervous system organization.
A threefold approach that combines gene-association studies with assessment of the epigenetic status of DD patients and analysis of the changes in animal models of depression, despite the limitations of such models ( 34 ), will enable researchers to identify the contributions of genetic, epigenetic, and environmental factors to different forms of DDs and to develop ways to reduce the risk of depression and to provide adequate treatment.
Author contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding. This study was supported by Russian Science Foundation (RSF) (project no. 16-15-10199).
Logo Left Content
Logo Right Content
Stanford University School of Medicine blog

Imagining virtual reality as a simple tool to treat depression
Some of the 17 million Americans afflicted with major depressive disorder each year may soon receive a surprising new prescription from their clinician: Have fun on a virtual reality device.
Engaging in activities that make you feel good may seem like overly simplistic advice, especially when directed at people with severe depression. But the science behind this idea, called "behavioral activation," is well established. Multiple studies have found that encouraging people to get outside, exercise, socialize, volunteer or immerse themselves in enjoyable activities in a prescribed, systematic way can help ease the symptoms of depression.
- Virtual reality helps people with hoarding disorder practice decluttering
Now, Stanford researchers have discovered that engaging in these behaviors within a virtual reality system may show just as much efficacy in treating depression as carrying them out in the real world. And for those depressed to a level that makes leaving the house a challenge, it could provide the benefits of getting outside -- and even motivate them to get out.

"People who might otherwise have barriers to getting treatment might be open to using this technology in their own homes," said Kim Bullock, MD , a clinical professor of psychiatry and behavioral sciences.
The study by Bullock's team, published in JMIR Mental Health , followed 26 people with major depressive disorder. Half were assigned traditional behavioral activation, and half used a virtual reality headset to participate in activities ranging from table tennis and mini-golf to touring foreign cities or attending shows. People in both groups saw their depression scores decrease by similar amounts.
"We've found that using virtual reality in an outpatient group of patients was both simple and efficacious in treating symptoms of depression," said Bullock, founder and director of Stanford's Neurobehavioral Clinic and Virtual Reality and Immersive Technologies (VRIT) program. "It can reduce the barriers to getting mental health treatment in a number of ways."
Bullock and her colleagues at the Stanford VRIT program have long studied the diverse ways to treat mental illnesses with virtual reality (VR) platforms, in which users donning headsets are immersed in simulated, three-dimensional environments.
Previous studies have examined how VR can be used to conduct therapy appointments, help people overcome anxieties and phobias, ease pain, learn social skills, and treat eating disorders and hoarding . But few research projects had focused on how to use the technology to treat anything as pervasive as major depressive disorder or other mood disorders
Depression impacts so many people right now, and we thought VR could have a large impact. Kim Bullock
"Depression impacts so many people right now, and we thought VR could have a large impact," Bullock said. "There can be significant barriers to behavioral activation in some patients -- they might be stuck in a hospital bed, or not have the means to access joyful activities or the motivation to leave their house. We started wondering whether simulated, pleasant activities might be a good first step for some people."
Bullock, along with clinical assistant professor Margot Paul, PsyD , first carried out a small feasibility study to see whether people with depression could use a VR headset with pre-loaded videos to engage in behavioral activation homework assigned by their therapist. After positive feedback from participants, the researchers conducted the randomized, controlled trial to test the efficacy of a more immersive and interactive VR approach.

The participants in the trial, all adults diagnosed with major depressive disorder who had not recently changed medications, met weekly with a clinical psychologist at Stanford who assigned them behavioral activation homework between sessions -- scheduling and committing to at least four pleasurable activities each week, either in virtual reality or real life.
Thirteen people in the study received a VR Meta Quest 2 headset as well as a list of potential activity ideas they could engage in using the headset, including games, travel videos, fitness classes, chat programs and education apps. The other 13 people were told to plan and partake in real life activities in a more typical fashion -- by going on outings in their community or socializing with friends.
After four weeks, both groups saw a significant decrease in their symptoms of depression and their depression rating on a widely used scale. Moreover, many people who had used the VR devices said the virtual activities had helped push them to get out of the house and be more involved in in-person activities.
"One of the most common pieces of feedback we got was that using the VR inspired people to get out and do things in the real world," Paul said. "These virtual activities got their motors running just enough to get out of bed."
- Stanford Medicine uses augmented reality for real-time data visualization during surgery
The only negative feedback pertained to learning how to set up the device, as well as the need for alerts or reminders to keep people accountable for engaging in the behavioral activation. Paul and Bullock have since developed a companion VR behavioral activation app that will help address some of these concerns.
The team says larger and longer-term studies are needed to find the best ways to administer virtual behavioral activation, as well as which patient populations might be best targeted with the VR treatment. They also think more efforts are needed to inform clinicians -- from therapists and psychologists to primary care doctors -- about how to prescribe VR behavioral activation appropriately.
These virtual activities got their motors running just enough to get out of bed. Margot Paul
But Paul, Bullock and their colleagues at Stanford VRIT believe the cost and ease of many VR platforms -- especially those that use mobile phones inserted into cheap cardboard headsets -- make it an easy treatment to scale up. They also believe the technology's relatability to a younger audience can only bring more openness to treating serious conditions like depression.
"As something that seems cool to young people, it serves not only to enhance but also de-stigmatize mental health treatments," Bullock said.
Illustration: Emily Moskal
More news about depression
- Serious talk about moods with bipolar disorder expert Po Wang
- Pilot study shows ketogenic diet improves severe mental illness
- Psychoactive drug ibogaine effectively treats traumatic brain injury in special ops military vets
- Human Neural Circuitry program seeks to investigate deepest mysteries of brain function, dysfunction
- Ketamine's effect on depression may hinge on hope
- Stanford Medicine-led research identifies a subtype of depression
- Researchers treat depression by reversing brain signals traveling the wrong way
- Stanford Medicine researchers find possible cause of depression after stroke
Popular posts

Why detecting the earliest biological signs of Parkinson’s disease is so crucial

Are long COVID sufferers falling through the cracks?
Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles and JavaScript.
- View all journals
- Explore content
- About the journal
- Publish with us
- Sign up for alerts
- Review Article
- Published: 29 December 2015
The role of inflammation in depression: from evolutionary imperative to modern treatment target
- Andrew H. Miller 1 &
- Charles L. Raison 2
Nature Reviews Immunology volume 16 , pages 22–34 ( 2016 ) Cite this article
88k Accesses
2066 Citations
631 Altmetric
Metrics details
- Adaptive immunity
- Inflammation
- Innate immunity
Across evolutionary time, inflammatory responses and depressive symptoms were part of an integrated adaptive response to pathogens that facilitated fighting infection, healing wounds and avoiding subsequent pathogen exposure in the pathogen-rich environments in which humans evolved. In the more sanitary environments of the modern world, the relationship between inflammatory pathways and the brain may drive depression and contribute to non-response to antidepressant medication.
Increased levels of inflammatory cytokines and induction of their signalling pathways as well as activation of different immune cell subsets has been detected in the brain and peripheral blood of a subgroup of patients with depression. C-reactive protein (CRP), tumour necrosis factor, interleukin-1β (IL-1β) and IL-6 appear to be the most reliably elevated inflammatory markers in the peripheral blood of subjects with depression.
Activation of the inflammasome by stress-induced, non-pathogenic stimuli, including damage-associated molecular patterns as well as microbial-associated molecular patterns elaborated from the gut microbiome, may drive peripheral inflammatory responses, which are then transmitted to the brain by trafficking of activated monocytes.
Inflammation impacts several neurotransmitter systems in the brain, including serotonin, dopamine and glutamate pathways, as well as the kynurenine pathway, which generates the neurotoxic metabolite quinolinic acid. Neuroimaging studies have demonstrated that disruption of neurotransmitter pathways is associated with inflammation-induced alterations in brain circuits that mediate motivation and motor activity as well as anxiety, arousal and alarm.
Activation of effector T cells during stress can prevent the development of depressive- and anxiety-like behaviour in mice. These effects may be mediated by the trafficking of effector T cells to the meningeal space where they produce IL-4, which supports anti-inflammatory responses while also stimulating the production of growth factors in the brain that support neural plasticity and resilience.
Studies in depression suggest that inflammatory biomarkers, such as CRP, can be used to enrich samples for anti-inflammatory clinical trials for depression that target inflammation-related symptoms such as anhedonia and anxiety, thereby supporting intelligent trial design. Though still in development, imaging of neuroinflammation will help establish a 'target' in the brain to further facilitate the testing of anti-inflammatory therapies for depression.
Crosstalk between inflammatory pathways and neurocircuits in the brain can lead to behavioural responses, such as avoidance and alarm, that are likely to have provided early humans with an evolutionary advantage in their interactions with pathogens and predators. However, in modern times, such interactions between inflammation and the brain appear to drive the development of depression and may contribute to non-responsiveness to current antidepressant therapies. Recent data have elucidated the mechanisms by which the innate and adaptive immune systems interact with neurotransmitters and neurocircuits to influence the risk for depression. Here, we detail our current understanding of these pathways and discuss the therapeutic potential of targeting the immune system to treat depression.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
195,33 € per year
only 16,28 € per issue
Rent or buy this article
Prices vary by article type
Prices may be subject to local taxes which are calculated during checkout
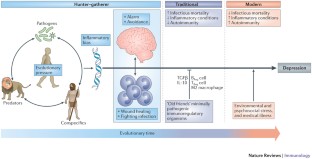
Similar content being viewed by others

Immune targets for therapeutic development in depression: towards precision medicine
Wayne C. Drevets, Gayle M. Wittenberg, … Husseini K. Manji

Molecular pathways of major depressive disorder converge on the synapse
Gabriel R. Fries, Valeria A. Saldana, … Theo Rein

Towards a multilevel model of major depression: genes, immuno-metabolic function, and cortico-striatal signaling
Elisabeth R. Paul, Lars Östman, … J. Paul Hamilton
Global Burden of Disease Study 2013 Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 386 , 743–800 (2015).
Rush, A. J. et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am. J. Psychiatry 163 , 1905–1917 (2006).
Article PubMed Google Scholar
Pace, T. W. et al. Increased stress-induced inflammatory responses in male patients with major depression and increased early life stress. Am. J. Psychiatry 163 , 1630–1633 (2006).
Bierhaus, A. et al. A mechanism converting psychosocial stress into mononuclear cell activation. Proc. Natl Acad. Sci. USA 100 , 1920–1925 (2003). This study is one of the first demonstrations that a psychological stressor could activate fundamental inflammatory signalling pathways (that is, NF-κB) in human peripheral blood mononuclear cells.
Article CAS PubMed Google Scholar
Aschbacher, K. et al. Maintenance of a positive outlook during acute stress protects against pro-inflammatory reactivity and future depressive symptoms. Brain Behav. Immun. 26 , 346–352 (2012).
Raison, C. L. & Miller, A. H. The evolutionary significance of depression in Pathogen Host Defense (PATHOS-D). Mol. Psychiatry 18 , 15–37 (2013). This theoretical treatise proposes that depression, rather than being a maladaptive response to psychosocial challenge, is the outgrowth of an evolutionary advantage provided by a crosstalk between the immune system and the brain to survive ancestral challenges from pathogens and predators.
Watson, P. J. & Andrews, P. W. Toward a revised evolutionary adaptationist analysis of depression: the social navigation hypothesis. J. Affect. Disord. 72 , 1–14 (2002).
Kinney, D. K. & Tanaka, M. An evolutionary hypothesis of depression and its symptoms, adaptive value, and risk factors. J. Nerv. Ment. Dis. 197 , 561–567 (2009).
Slavich, G. M. & Irwin, M. R. From stress to inflammation and major depressive disorder: a social signal transduction theory of depression. Psychol. Bull. 140 , 774–815 (2014).
Article PubMed PubMed Central Google Scholar
Seedat, S. et al. Cross-national associations between gender and mental disorders in the World Health Organization World Mental Health Surveys. Arch. Gen. Psychiatry 66 , 785–795 (2009).
Moieni, M. et al. Sex differences in depressive and socioemotional responses to an inflammatory challenge: implications for sex differences in depression. Neuropsychopharmacology 40 , 1709–1716 (2015).
Article CAS PubMed PubMed Central Google Scholar
Udina, M. et al. Interferon-induced depression in chronic hepatitis C: a systematic review and meta-analysis. J. Clin. Psychiatry 73 , 1128–1138 (2012).
Raison, C. L., Lowry, C. A. & Rook, G. A. Inflammation, sanitation, and consternation: loss of contact with coevolved, tolerogenic microorganisms and the pathophysiology and treatment of major depression. Arch. Gen. Psychiatry 67 , 1211–1224 (2010).
Rook, G. A., Lowry, C. A. & Raison, C. L. Hygiene and other early childhood influences on the subsequent function of the immune system. Brain Res. 1617 , 47–62 (2015).
Yirmiya, R. et al. Illness, cytokines, and depression. Ann. NY Acad. Sci. 917 , 478–487 (2000).
Miller, A. H., Maletic, V. & Raison, C. L. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 65 , 732–741 (2009).
Maes, M. Major depression and activation of the inflammatory response system. Adv. Exp. Med. Biol. 461 , 25–46 (1999).
Brambilla, P. et al. Increased M1/decreased M2 signature and signs of Th1/Th2 shift in chronic patients with bipolar disorder, but not in those with schizophrenia. Transl Psychiatry 4 , e406 (2014).
Drago, A., Crisafulli, C., Calabro, M. & Serretti, A. Enrichment pathway analysis. The inflammatory genetic background in bipolar disorder. J. Affect Disord. 179 , 88–94 (2015).
Mostafavi, S. et al. Type I interferon signaling genes in recurrent major depression: increased expression detected by whole-blood RNA sequencing. Mol. Psychiatry 19 , 1267–1274 (2013).
Maes, M. Evidence for an immune response in major depression: a review and hypothesis. Prog. Neuropsychopharmacol. Biol. Psychiatry 19 , 11–38 (1995). In contrast to previous theories that primarily focused on reduced T cell responses to mitogenic stimuli, this is one of the first papers to posit that an activated immune system may have a role in the aetiology of depression.
Bufalino, C., Hepgul, N., Aguglia, E. & Pariante, C. M. The role of immune genes in the association between depression and inflammation: a review of recent clinical studies. Brain Behav. Immun. 31 , 31–47 (2012).
Capuron, L. et al. Neurobehavioral effects of interferon-α in cancer patients: phenomenology and paroxetine responsiveness of symptom dimensions. Neuropsychopharmacology 26 , 643–652 (2002).
Reichenberg, A. et al. Cytokine-associated emotional and cognitive disturbances in humans. Arch. Gen. Psychiatry 58 , 445–452 (2001).
Bonaccorso, S. et al. Increased depressive ratings in patients with hepatitis C receiving interferon-α-based immunotherapy are related to interferon-α-induced changes in the serotonergic system. J. Clin. Psychopharmacol. 22 , 86–90 (2002).
Harrison, N. A. et al. Inflammation causes mood changes through alterations in subgenual cingulate activity and mesolimbic connectivity. Biol. Psychiatry 66 , 407–414 (2009).
Tyring, S. et al. Etanercept and clinical outcomes, fatigue, and depression in psoriasis: double-blind placebo-controlled randomised phase III trial. Lancet 367 , 29–35 (2006).
Abbott, R. et al. Tumour necrosis factor-α inhibitor therapy in chronic physical illness: a systematic review and meta-analysis of the effect on depression and anxiety. J. Psychosom. Res. 79 , 175–84 (2015).
Kohler, O. et al. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: a systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry 71 , 1381–1391 (2014).
Raison, C. L. et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry 70 , 31–41 (2013). This report describes the results of the first double-blind, placebo-controlled trial of a monoclonal antibody against TNF to treat major depression, indicating that only patients with depression that have high levels of inflammation respond to cytokine antagonism.
Miller, A. H. & Raison, C. L. Are anti-inflammatory therapies viable treatments for psychiatric disorders?: where the rubber meets the road. JAMA Psychiatry 72 , 527–528 (2015).
Michopoulos, V. et al. Association of CRP genetic variation and CRP level with elevated PTSD symptoms and physiological responses in a civilian population with high levels of trauma. Am. J. Psychiatry 172 , 353–362 (2015).
Fernandes, B. S. et al. C-reactive protein is increased in schizophrenia but is not altered by antipsychotics: meta-analysis and implications. Mol. Psychiatry http://dx.doi.org/10.1038/mp.2015.87 (2015).
Morris, S. E. & Cuthbert, B. N. Research Domain Criteria: cognitive systems, neural circuits, and dimensions of behavior. Dialogues Clin. Neurosci. 14 , 29–37 (2012).
PubMed PubMed Central Google Scholar
Miller, A. H., Haroon, E., Raison, C. L. & Felger, J. C. Cytokine targets in the brain: impact on neurotransmitters and neurocircuits. Depress. Anxiety 30 , 297–306 (2013).
Cattaneo, A. et al. Candidate genes expression profile associated with antidepressants response in the GENDEP study: differentiating between baseline 'predictors' and longitudinal 'targets'. Neuropsychopharmacology 38 , 377–385 (2013).
Eurelings, L. S., Richard, E., Eikelenboom, P., van Gool, W. A. & Moll van Charante, E. P. Low-grade inflammation differentiates between symptoms of apathy and depression in community-dwelling older individuals. Int. Psychogeriatr. 27 , 639–647 (2015).
Pearson, T. A. et al. Markers of inflammation and cardiovascular disease: application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation 107 , 499–511 (2003).
Irwin, M. R. & Cole, S. W. Reciprocal regulation of the neural and innate immune systems. Nat. Rev. Immunol. 11 , 625–632 (2011).
Iwata, M., Ota, K. T. & Duman, R. S. The inflammasome: pathways linking psychological stress, depression, and systemic illnesses. Brain Behav. Immun. 31 , 105–114 (2013).
Strowig, T., Henao-Mejia, J., Elinav, E. & Flavell, R. Inflammasomes in health and disease. Nature 481 , 278–286 (2012).
Fleshner, M. Stress-evoked sterile inflammation, danger associated molecular patterns (DAMPs), microbial associated molecular patterns (MAMPs) and the inflammasome. Brain Behav. Immun. 27 , 1–7 (2013).
Cox, S. S. et al. Adrenergic and glucocorticoid modulation of the sterile inflammatory response. Brain Behav. Immun. 36 , 183–192 (2014).
Pan, Y., Chen, X. Y., Zhang, Q. Y. & Kong, L. D. Microglial NLRP3 inflammasome activation mediates IL-1β-related inflammation in prefrontal cortex of depressive rats. Brain Behav. Immun. 41 , 90–100 (2014).
Zhang, Y. et al. NLRP3 inflammasome mediates chronic mild stress-induced depression in mice via neuroinflammation. Int. J. Neuropsychopharmacol. 18 , pii: pyv006 (2015).
Article CAS Google Scholar
Paugh, S. W. et al. NALP3 inflammasome upregulation and CASP1 cleavage of the glucocorticoid receptor cause glucocorticoid resistance in leukemia cells. Nat. Genet. 47 , 607–614 (2015).
Rhen, T. & Cidlowski, J. A. Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N. Engl. J. Med. 353 , 1711–1723 (2005).
Raison, C. L. & Miller, A. H. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am. J. Psychiatry 160 , 1554–1565 (2003).
Pace, T. W., Hu, F. & Miller, A. H. Cytokine-effects on glucocorticoid receptor function: relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav. Immun. 21 , 9–19 (2007).
Alcocer-Gomez, E. et al. NLRP3 inflammasome is activated in mononuclear blood cells from patients with major depressive disorder. Brain Behav. Immun. 36 , 111–117 (2014). This paper provides the first indication that activation of the inflammasome may contribute to elevated levels of inflammatory cytokines such as IL-1β and IL-18 in major depression, consistent with studies in laboratory animal models of depression that demonstrated that inhibition of NLRP3 can block the development of stress-induced depressive-like behaviour.
Stertz, L. et al. Damage-associated molecular patterns and immune activation in bipolar disorder. Acta Psychiatr. Scand. 132 , 211–217 (2015).
Rawdin, B. J. et al. Dysregulated relationship of inflammation and oxidative stress in major depression. Brain Behav. Immun. 31 , 143–152 (2013).
Maes, M., Galecki, P., Chang, Y. S. & Berk, M. A review on the oxidative and nitrosative stress (O&NS) pathways in major depression and their possible contribution to the (neuro)degenerative processes in that illness. Prog. Neuropsychopharmacol. Biol. Psychiatry 35 , 676–692 (2011).
Mayer, E. A., Knight, R., Mazmanian, S. K., Cryan, J. F. & Tillisch, K. Gut microbes and the brain: paradigm shift in neuroscience. J. Neurosci. 34 , 15490–15496 (2014).
Maslanik, T. et al. Commensal bacteria and MAMPs are necessary for stress-induced increases in IL-1β and IL-18 but not IL-6, IL-10 or MCP-1. PLoS ONE 7 , e50636 (2012).
Lyte, M., Vulchanova, L. & Brown, D. R. Stress at the intestinal surface: catecholamines and mucosa-bacteria interactions. Cell Tissue Res. 343 , 23–32 (2011).
Rao, J. S., Harry, G. J., Rapoport, S. I. & Kim, H. W. Increased excitotoxicity and neuroinflammatory markers in postmortem frontal cortex from bipolar disorder patients. Mol. Psychiatry 15 , 384–392 (2010).
Steiner, J. et al. Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J. Psychiatr. Res. 42 , 151–157 (2008).
Torres-Platas, S. G., Cruceanu, C., Chen, G. G., Turecki, G. & Mechawar, N. Evidence for increased microglial priming and macrophage recruitment in the dorsal anterior cingulate white matter of depressed suicides. Brain Behav. Immun. 42 , 50–59 (2014). This study provides some of the most compelling evidence that neuroinflammation occurs in major depression by demonstrating that monocytes traffic to the brain of patients with depressive symptoms and assume a perivascular localization in association with chemoattractant molecules such as CCL2, which has been shown to attract monocytes to the brain in animal models of stress.
Nagy, C. et al. Astrocytic abnormalities and global DNA methylation patterns in depression and suicide. Mol. Psychiatry 20 , 320–328 (2015).
Setiawan, E. et al. Role of translocator protein density, a marker of neuroinflammation, in the brain during major depressive episodes. JAMA Psychiatry 72 , 268–275 (2015).
Hannestad, J. et al. The neuroinflammation marker translocator protein is not elevated in individuals with mild-to-moderate depression: a [ 11 C]PBR28 PET study. Brain Behav. Immun. 33 , 131–138 (2013).
Sandiego, C. M. et al. Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc. Natl Acad. Sci. USA 112 , 12468–12473 (2015).
Quan, N. & Banks, W. A. Brain-immune communication pathways. Brain Behav. Immun. 21 , 727–735 (2007).
D'Mello, C., Le, T. & Swain, M. G. Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factor-α signaling during peripheral organ inflammation. J. Neurosci. 29 , 2089–2102 (2009).
Hennessy, E., Griffin, E. W. & Cunningham, C. Astrocytes are primed by chronic neurodegeneration to produce exaggerated chemokine and cell infiltration responses to acute stimulation with the cytokines IL-1β and TNF-α. J. Neurosci. 35 , 8411–8422 (2015).
Wohleb, E. S. et al. β-adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J. Neurosci. 31 , 6277–6288 (2011).
Wohleb, E. S., Powell, N. D., Godbout, J. P. & Sheridan, J. F. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J. Neurosci. 33 , 13820–13833 (2013). References 67 and 68 present a series of experiments that demonstrate a cellular pathway by which cytokines signals can be transmitted to the brain via trafficking of monocytes from the bone marrow to the brain parenchyma, a process mediated by catecholamines and CCL2.
Krugel, U., Fischer, J., Radicke, S., Sack, U. & Himmerich, H. Antidepressant effects of TNF-α blockade in an animal model of depression. J. Psychiatr. Res. 47 , 611–616 (2013).
Arends, S. et al. Baseline predictors of response and discontinuation of tumor necrosis factor-α blocking therapy in ankylosing spondylitis: a prospective longitudinal observational cohort study. Arthritis Res. Ther. 13 , R94 (2011).
Gillespie, C. F., Garlow, S. J., Binder, E. B., Schatzberg, A. F. & Nemeroff, C. B. in Textbook of Psychopharmacology (eds Schatzberg, A. F. & Nemeroff, C. B.) 903–944 (America Psychiatric Publishing, 2009).
Google Scholar
Zhu, C. B. et al. Interleukin-1 receptor activation by systemic lipopolysaccharide induces behavioral despair linked to MAPK regulation of CNS serotonin transporters. Neuropsychopharmacology 35 , 2510–2520 (2010).
Neurauter, G. et al. Chronic immune stimulation correlates with reduced phenylalanine turnover. Curr. Drug Metab. 9 , 622–627 (2008).
Felger, J. C. et al. Tyrosine metabolism during interferon-α administration: association with fatigue and CSF dopamine concentrations. Brain Behav. Immun. 31 , 153–160 (2013).
Maes, M., Leonard, B. E., Myint, A. M., Kubera, M. & Verkerk, R. The new '5-HT' hypothesis of depression: cell-mediated immune activation induces indoleamine 2,3-dioxygenase, which leads to lower plasma tryptophan and an increased synthesis of detrimental tryptophan catabolites (TRYCATs), both of which contribute to the onset of depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 35 , 702–721 (2011).
Raison, C. L. et al. CSF concentrations of brain tryptophan and kynurenines during immune stimulation with IFN-α: relationship to CNS immune responses and depression. Mol. Psychiatry 15 , 393–403 (2010).
Steiner, J. et al. Severe depression is associated with increased microglial quinolinic acid in subregions of the anterior cingulate gyrus: evidence for an immune-modulated glutamatergic neurotransmission? J. Neuroinflamm. 8 , 94 (2011).
Tavares, R. G. et al. Quinolinic acid stimulates synaptosomal glutamate release and inhibits glutamate uptake into astrocytes. Neurochem. Int. 40 , 621–627 (2002).
Tilleux, S. & Hermans, E. Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J. Neurosci. Res. 85 , 2059–2070 (2007).
Hardingham, G. E., Fukunaga, Y. & Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 5 , 405–414 (2002).
Koo, J. W., Russo, S. J., Ferguson, D., Nestler, N. J. & Duman, R. S. Nuclear factor-κB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc. Natl Acad. Sci. USA 107 , 2669–2674 (2010).
Goshen, I. et al. Brain interleukin-1 mediates chronic stress-induced depression in mice via adrenocortical activation and hippocampal neurogenesis suppression. Mol. Psychiatry 13 , 717–728 (2008).
Haroon, E. et al. IFN-α-induced cortical and subcortical glutamate changes assessed by magnetic resonance spectroscopy. Neuropsychopharmacology 39 , 1777–1785 (2014).
Haroon, E. et al. Conceptual convergence:increased inflammation is associated with increased basal ganglia glutamate in patients with major depression. Mol. Psychiatry http://dx.doi.org/10.1038/mp.2015.206 (2015).
Walker, A. K. et al. NMDA receptor blockade by ketamine abrogates lipopolysaccharide-induced depressive-like behavior in C57BL/6J mice. Neuropsychopharmacology 38 , 1609–1616 (2013).
O'Connor, J. C. et al. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol. Psychiatry 14 , 511–522 (2009).
Duman, R. S. & Monteggia, L. M. A neurotrophic model for stress-related mood disorders. Biol. Psychiatry 59 , 1116–1127 (2006).
Hodes, G. E. et al. Individual differences in the peripheral immune system promote resilience versus susceptibility to social stress. Proc. Natl Acad. Sci. USA 111 , 16136–16141 (2014). This paper gives compelling evidence in laboratory animals that individual differences in the behavioural response to a social stressor is mediated by individual differences in the production of the inflammatory cytokine IL-6, suggesting that genetic and/or environmental factors that regulate inflammatory responses can determine stress-induced depressive-like behaviour.
Capuron, L. et al. Dopaminergic mechanisms of reduced basal ganglia responses to hedonic reward during interferon-α administration. Arch. Gen. Psychiatry 69 , 1044–1053 (2012). Using multimodal neuroimaging, this report provides an integration of the impact of inflammatory cytokines on reward pathways and dopamine metabolism in the basal ganglia that leads to alterations in behaviours relevant to motivation and ultimately anhedonia — a core symptom of depression.
Eisenberger, N. I. et al. Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol. Psychiatry 68 , 748–754 (2010).
Felger, J. C. et al. Chronic interferon-α decreases dopamine 2 receptor binding and striatal dopamine release in association with anhedonia-like behavior in nonhuman primates. Neuropsychopharmacology 38 , 2179–2187 (2013).
Dowell, N. G. et al. Acute changes in striatal microstructure predict the development of interferon-α induced fatigue. Biol. Psychiatry http://dx.doi.org/10.1016/j.biopsych.2015.05.015 (2015).
Harrison, N. A., Cercignani, M., Voon, V. & Critchley, H. D. Effects of inflammation on hippocampus and substantia nigra responses to novelty in healthy human participants. Neuropsychopharmacology 40 , 831–838 (2015).
Harrison, N. A. et al. A neuro-computational account of how inflammation enhances sensitivity to punishments versus rewards. Biol. Psychiatry http://dx.doi.org/10.1016/j.biopsych.2015.07.018 (2015).
Felger, J. C. et al. Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol. Psychiatry http://dx.doi.org/10.1038/mp.2015.168 (2015). This neuroimaging study is the first to demonstrate that the impact of immune stimuli (for example, IFN α, typhoid vaccination and endotoxin) on the brains of otherwise non-depressed individuals extended to patients with depression, whose increased inflammation was found to reduce functional connectivity in reward-related neurocircuitry leading to anhedonia, a core symptom of depression.
Harrison, N. A. et al. Neural origins of human sickness in interoceptive responses to inflammation. Biol. Psychiatry 66 , 415–422 (2009).
Slavich, G. M., Way, B. M., Eisenberger, N. I. & Taylor, S. E. Neural sensitivity to social rejection is associated with inflammatory responses to social stress. Proc. Natl Acad. Sci. USA 107 , 14817–14822 (2010). This manuscript provides one of the first and most detailed descriptions of the relationship between stress-induced activation of inflammatory responses and sensitivity to a psychosocial stressor, identifying key brain regions such as the dACC as a target of cytokines, leading to anxiety, arousal and alarm.
Eisenberger, N. I. & Lieberman, M. D. Why rejection hurts: a common neural alarm system for physical and social pain. Trends Cogn. Sci. 8 , 294–300 (2004).
Muscatell, K. A. et al. Greater amygdala activity and dorsomedial prefrontal-amygdala coupling are associated with enhanced inflammatory responses to stress. Brain Behav. Immun. 43 , 46–53 (2015).
Gimeno, D. et al. Associations of C-reactive protein and interleukin-6 with cognitive symptoms of depression: 12-year follow-up of the Whitehall II study. Psychol. Med. 39 , 413–423 (2009).
Au, B., Smith, K. J., Gariepy, G. & Schmitz, N. The longitudinal associations between C-reactive protein and depressive symptoms: evidence from the English Longitudinal Study of Ageing (ELSA). Int. J. Geriatr. Psychiatry 30 , 976–984 (2015).
Duivis, H. E. et al. Depressive symptoms, health behaviors, and subsequent inflammation in patients with coronary heart disease: prospective findings from the heart and soul study. Am. J. Psychiatry 168 , 913–920 (2011).
Baumeister, D., Akhtar, R., Ciufolini, S., Pariante, C. M. & Mondelli, V. Childhood trauma and adulthood inflammation: a meta-analysis of peripheral C-reactive protein, interleukin-6 and tumour necrosis factor-α. Mol. Psychiatry http://dx.doi.org/10.1038/mp.2015.67 (2015).
Danese, A., Pariante, C. M., Caspi, A., Taylor, A. & Poulton, R. Childhood maltreatment predicts adult inflammation in a life-course study. Proc. Natl Acad. Sci. USA 104 , 1319–1324 (2007).
Tartter, M., Hammen, C., Bower, J. E., Brennan, P. A. & Cole, S. Effects of chronic interpersonal stress exposure on depressive symptoms are moderated by genetic variation at IL6 and IL1β in youth. Brain Behav. Immun. 46 , 104–111 (2015).
Klengel, T. et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat. Neurosci. 16 , 33–41 (2013).
Miller, G. E. et al. A functional genomic fingerprint of chronic stress in humans: blunted glucocorticoid and increased NF-κB signaling. Biol. Psychiatry 64 , 266–272 (2008).
Brachman, R. A., Lehmann, M. L., Maric, D. & Herkenham, M. Lymphocytes from chronically stressed mice confer antidepressant-like effects to naive mice. J. Neurosci. 35 , 1530–1538 (2015).
Lewitus, G. M., Cohen, H. & Schwartz, M. Reducing post-traumatic anxiety by immunization. Brain Behav. Immun. 22 , 1108–1114 (2008).
Lewitus, G. M. et al. Vaccination as a novel approach for treating depressive behavior. Biol. Psychiatry 65 , 283–288 (2009). References 109 and 110 provide the first evidence that engaging T cells to traffic to the brain during stress can block depressive- and anxiety-like behaviour and induce growth factors and neurogenesis in the brain, representing an entirely novel approach to the treatment of depression that takes advantage of the capacity of T cells to support neuronal integrity.
Derecki, N. C. et al. Regulation of learning and memory by meningeal immunity: a key role for IL-4. J. Exp. Med. 207 , 1067–1080 (2010).
Louveau, A. et al. Structural and functional features of central nervous system lymphatic vessels. Nature 523 , 337–341 (2015).
Kim, S. J. et al. CD4 + CD25 + regulatory T cell depletion modulates anxiety and depression-like behaviors in mice. PLoS ONE 7 , e42054 (2012).
Rosas-Ballina, M. et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 334 , 98–101 (2011).
Bauer, M. E. et al. Dexamethasone-induced effects on lymphocyte distribution and expression of adhesion molecules in treatment-resistant depression. Psychiatry Res. 113 , 1–15 (2002).
Wang, X., Wu, H. & Miller, A. H. Interleukin 1α (IL-1α) induced activation of p38 mitogen-activated protein kinase inhibits glucocorticoid receptor function. Mol. Psychiatry 9 , 65–75 (2004).
Wei, J., Zhang, M. & Zhou, J. Myeloid-derived suppressor cells in major depression patients suppress T-cell responses through the production of reactive oxygen species. Psychiatry Res. 228 , 695–701 (2015).
van Deventer, H. W. et al. The inflammasome component NLRP3 impairs antitumor vaccine by enhancing the accumulation of tumor-associated myeloid-derived suppressor cells. Cancer Res. 70 , 10161–10169 (2010).
Li, Y. et al. Altered expression of CD4 + CD25 + regulatory T cells and its 5-HT1a receptor in patients with major depression disorder. J. Affect. Disord. 124 , 68–75 (2010).
Williamson, L. L. et al. Got worms? Perinatal exposure to helminths prevents persistent immune sensitization and cognitive dysfunction induced by early-life infection. Brain Behav. Immun. (2015).
Olofsson, P. S., Rosas-Ballina, M., Levine, Y. A. & Tracey, K. J. Rethinking inflammation: neural circuits in the regulation of immunity. Immunol. Rev. 248 , 188–204 (2012).
Wang, Y., Chen, X., Cao, W. & Shi, Y. Plasticity of mesenchymal stem cells in immunomodulation: pathological and therapeutic implications. Nat. Immunol. 15 , 1009–1016 (2014).
Warner-Schmidt, J. L., Vanover, K. E., Chen, E. Y., Marshall, J. J. & Greengard, P. Antidepressant effects of selective serotonin reuptake inhibitors (SSRIs) are attenuated by antiinflammatory drugs in mice and humans. Proc. Natl Acad. Sci. USA 108 , 9262–9267 (2011).
Gurven, M. & Kaplan, H. Longevity among hunter-gatherers: a cross-cultural examination. Popul. Dev. Rev. 33 , 321–365 (2007).
Article Google Scholar
Fumagalli, M. et al. Signatures of environmental genetic adaptation pinpoint pathogens as the main selective pressure through human evolution. PLoS Genet. 7 , e1002355 (2011).
Kuningas, M. et al. Selection for genetic variation inducing pro-inflammatory responses under adverse environmental conditions in a Ghanaian population. PLoS ONE 4 , e7795 (2009).
Ratcliffe, M. A bad case of the flu?: the comparative phenomenology of depression and somatic illness. J. Conscious. Studies 20 , 198–218 (2013).
Marin, I. & Kipnis, J. Learning and memory... and the immune system. Learn. Mem. 20 , 601–606 (2013).
Mehta, D. et al. Transcriptional signatures related to glucose and lipid metabolism predict treatment response to the tumor necrosis factor antagonist infliximab in patients with treatment-resistant depression. Brain Behav. Immun. 31 , 205–215 (2013).
Download references
Author information
Authors and affiliations.
Emory University School of Medicine, Winship Cancer Institute, Atlanta, 30322, Georgia, USA
Andrew H. Miller
School of Human Ecology, University of Wisconsin–Madison, Madison, 53706, Wisconsin, USA
Charles L. Raison
You can also search for this author in PubMed Google Scholar
Corresponding author
Correspondence to Andrew H. Miller .
Ethics declarations
Competing interests.
The authors declare no competing financial interests.
PowerPoint slides
Powerpoint slide for fig. 1, powerpoint slide for fig. 2, powerpoint slide for fig. 3.
Members of the same species.
An adaptive response to illness, often precipitated by infection, that includes social withdrawal, decreased appetite, lethargy, impaired concentration, depressed mood, irritability, muscle aches and pain, and fever. This syndrome is believed to prioritize shifting of energy resources to fighting infection and wound healing.
A lack of interest in usually pleasurable activities that represents a decrease in motivation, which can either represent a decrease in the response to reward or in the willingness to expend effort to obtain reward.
A clinical syndrome of depression characterized by the primary symptoms of depressed mood and anhedonia, and diagnosed using criteria set forth by the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition.
A model of depression that entails repeated exposure to a conspecific animal screened for aggressive behaviour. The animals are placed together in the same cage where they are exposed to brief bouts of defeat lasting 5–10 minutes daily typically for 6–10 days.
A heterogeneous population of cells of myeloid origin that rapidly expands during inflammation and can potently suppress T cell responses. They are now being explored as potential therapeutic targets to inhibit immune responses in autoimmune disease or transplant rejection.
A theoretical framework that suggests that cytokines have a primary role in alterations of neurotransmitter metabolism, neuroendocrine function, neuroplasticity, neurocircuitry and behaviour in a subgroup of patients with depression and increased inflammation.
Rights and permissions
Reprints and permissions
About this article
Cite this article.
Miller, A., Raison, C. The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol 16 , 22–34 (2016). https://doi.org/10.1038/nri.2015.5
Download citation
Published : 29 December 2015
Issue Date : January 2016
DOI : https://doi.org/10.1038/nri.2015.5
Share this article
Anyone you share the following link with will be able to read this content:
Sorry, a shareable link is not currently available for this article.
Provided by the Springer Nature SharedIt content-sharing initiative
This article is cited by
Dose-response associations of maternal prenatal noise exposure duration with antepartum depression status.
- Deliang Wen
BMC Pregnancy and Childbirth (2024)
The quest for a biological phenotype of adolescent non-suicidal self-injury: a machine-learning approach
- Ines Mürner-Lavanchy
- Julian Koenig
- Michael Kaess
Translational Psychiatry (2024)
Evaluation of serum interleukin-12 and interleukin-4 as potential biomarkers for the diagnosis of major depressive disorder
- Nisat Sarmin
- A. S. M. Roknuzzaman
- Zobaer Al Mahmud
Scientific Reports (2024)
Elevated body temperature is associated with depressive symptoms: results from the TemPredict Study
- Ashley E. Mason
- Patrick Kasl
- Benjamin L. Smarr
Bad Feelings, Best Explanations: In Defence of the Propitiousness Theory of the Low Mood System
- James Turner
Erkenntnis (2024)
Quick links
- Explore articles by subject
- Guide to authors
- Editorial policies
Sign up for the Nature Briefing newsletter — what matters in science, free to your inbox daily.

IMAGES
VIDEO
COMMENTS
Worldwide, the incidence of major depressive disorder (MDD) is increasing annually, resulting in greater economic and social burdens. Moreover, the pathological mechanisms of MDD and the ...
This hypothesis is the basis for the application of monoamine oxidase inhibitors as antidepressant medications in the management of depressive illnesses, ... Thus, there is a strong evidence base for genetics underlying the pathophysiologic process of depression as a disease entity. 2.3. Environmental stress factors.
1. Introduction. Major depressive disorder (MDD) is a heterogeneous disease that affects one out of five individuals in their lifetime and is the leading cause of disability worldwide [].The symptoms of MDD are associated with structural and neurochemical deficits in the corticolimbic brain regions [2,3,4].The behavioral symptoms of depression are extensive, covering emotional, motivational ...
Depression can start early in the lifecourse and, if it remains unmanaged, may increase the risk for substance abuse, chronic conditions, such as cardiovascular disease, and premature mortality [4,5,6,7,8]. Treatment for depression exists, such as pharmacotherapy, cognitive behavioural therapy, and other modalities.
Abstract. Major depression is a serious disorder of enormous sociological and clinical relevance. The discovery of antidepressant drugs in the 1950s led to the first biochemical hypothesis of depression, which suggested that an impairment in central monoaminergic function was the major lesion underlying the disorder.
It has long been noted by clinicians that patients suffering from various endocrine disorders (e.g. Cushing's disease) often develop depressive symptoms (Cowen, 2015). In turn, patients with depression have been observed to exhibit a number of abnormalities in the hypothalamic-pituitary-adrenal (HPA) axis, most notably subtle signs of ...
Understanding depression pathophysiology is challenging because varying depression symptomatology cannot be explained by single hypothesis. • Pathophysiologic mechanisms include: monoamine hypothesis, genetic, environmental, immunologic, endocrine factors and neurogenesis. The Hypothalamo-Pituitary-Adrenal axis is the major neurobiological link between these factors and the development of ...
Depression is a common psychiatric disorder and a leading cause of disability worldwide. Here we conducted a genome-wide association study meta-analysis of six datasets, including >1.3 million ...
Abstract. Hypotheses about the pathophysiology of depression have evolved over time. This chapter covers the most important findings in this regard. First, the classical monoamine hypothesis posited that depression is caused by an alteration in levels of one or more of the monoamines: serotonin, norepinephrine, and dopamine.
The monoamine hypothesis of depression postulates a deficiency in serotonin or norepinephrine neurotransmission in the brain. ... both depression and cardiovascular disease could be examples of an ...
These considerations sustain the mitochondrial hypothesis of depression, which states that impaired energy metabolism of brain cells is involved in the pathophysiology of mood disorders and in the effects of antidepressants and mood stabiliser drugs. ... Maes M (2011) Depression is an inflammatory disease, but cell-mediated immune activation is ...
As a consequence, antidepressant treatments, including psychological and biological approaches, should be tailored for individual patients and disease states. Individual depression hypotheses based on neurobiological knowledge are discussed in terms of their interest to both clinicians in daily practice and clinical researchers developing novel ...
Abstract. Depression has become one of the most prevalent neuropsychiatric disorders characterized by anhedonia, anxiety, pessimism, or even suicidal thoughts. Receptor theory has been pointed out to explain the pathogenesis of depression, while it is still subject to debate. Additionally, gene abnormality accounts for nearly 40-50% of ...
Furthermore, we propose a new hypothesis for LLD, the age-by-disease interaction hypothesis, which posits that the clinical presentation of LLD is the integrated output of specific biologic processes that are pushed in LLD-promoting directions by changes in gene expression naturally occurring in the brain during aging.
The cytokine hypothesis and depressive symptoms. Systemic exposure to inflammatory challenges, such as lipopolysaccharide (LPS), not only causes a systemic inflammation, but also induces a central neuroinflammation, reflected by activation of brain microglia with a chronically elevated production of pro-inflammatory mediators, such as TNFα, which may remain elevated for 10 months (Qin et al ...
A model of depression as a disease is incomplete and misleading. It leads to suboptimal treatment choices. The "separation distress hypothesis" is an alternative model. It is an integrated, causal ...
Elevated levels of IL-6 in patients with COVID-19 infection. The world-wide effect of the coronavirus disease 2019 (COVID-19) pandemic is enormous and is not solely limited to the increased mortality and morbidity rates, but also extends into the mental health of the global population [].Considerable amount of clinical data is emerging regarding the manifestation of depression in patients ...
These observations led to the pharmacologically most relevant theory of depression, referred to as the monoamine-deficiency hypothesis. The monoamine-deficiency theory posits that the underlying pathophysiological basis of depression is a depletion of the neurotransmitters serotonin, norepinephrine or dopamine in the central nervous system.
By: Bill Snyder. Coronary artery disease and major depression may be genetically linked via inflammatory pathways to an increased risk for cardiomyopathy, a degenerative heart muscle disease, researchers at Vanderbilt University Medical Center and Massachusetts General Hospital have found. Their report, published April 5 in the journal Nature ...
Credit: Nature Mental Health (2024). DOI: 10.1038/s44220-024-00219-z. Coronary artery disease and major depression may be genetically linked via inflammatory pathways to an increased risk for ...
Coronary artery disease and major depression may be genetically linked via inflammatory pathways to an increased risk for cardiomyopathy, a degenerative heart muscle disease, researchers have found.
Depression is one of the most common mental health disorders in the US. About 18% of American adults - more than 1 in 6 - say they are depressed or receiving treatment for depression, a 2023 ...
Parkinson's disease (PD) is among the most prevalent neurodegenerative diseases, and approximately one third of patients with PD are estimated to have depression. Paraquat (PQ) exposure is an important environmental risk factor for PD. In this study, we established a mouse model of PQ-induced PD with depression to comprehensively investigate cellular heterogeneity and the mechanisms underlying ...
Major depressive disease (MDD), schizophrenia (SCZ), and bipolar disorder (BD) are common psychiatric disorders, and their relationship with thyroid cancer has been of great interest. This study aimed to investigate the potential causal effects of MDD, SCZ, BD, and thyroid cancer. We used publicly available summary statistics from large-scale genome-wide association studies to select genetic ...
The monoamine hypothesis of depression, the first theory historically, was proposed by Joseph Schildkraut in the 1960s. ... This table focuses mainly on those studies that analyzed primarily the risk of depression as a disease and not the endophenotypes (e.g., clinical onset age, severity of particular symptoms, patients' responses to therapy). ...
Now, Stanford researchers have discovered that engaging in these behaviors within a virtual reality system may show just as much efficacy in treating depression as carrying them out in the real world. And for those depressed to a level that makes leaving the house a challenge, it could provide the benefits of getting outside -- and even ...
ATLANTA — Younger women with a diagnosis of anxiety or depression are at markedly higher risk for developing conditions that increase CVD risk, such as hypertension, hyperlipidemia and ...
The prevalence of autoimmune, allergic and inflammatory diseases has markedly increased in the past 100 years, ... The new '5-HT' hypothesis of depression: cell-mediated immune activation induces ...